应用化学
2022, 39 (
):
1138-1146.
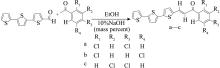
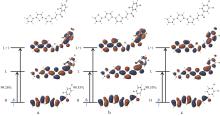
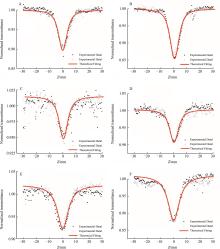
以α-三联噻吩甲醛为原料,与3种二氯苯乙酮发生Claisen-Schmidt缩合反应,合成了3种含有三联噻吩基的查尔酮衍生物:1-(α-三联噻吩-2-基)-3-(2,4-二氯苯基)丙烯酮(a)、1-(2,5-二氯苯基)-3-(α-三联噻吩-2-基)丙烯酮(b)和1-(3,4-二氯苯基)-3-(α-三联噻吩-2-基)丙烯酮(c)。借助核磁共振波谱仪(1H NMR、13C NMR)和液-质联用谱仪(LC-MS)对其结构进行了表征;采用Z-扫描技术(600 nm,180 fs)测定了3个化合物的非线性光学吸收性能;运用含时密度泛函理论(TD-DFT)方法计算了它们的极化率(α0)、静态第一超极化率(β0)、振子强度(f0)、跃迁能(ΔE)、基态和最主要激发态之间的偶极矩差(Δμ)、最主要激发态的主要组成、最高占据分子轨道(HOMO)和最低空分子轨道(LUMO)之间的能隙,同时测定了它们的线性光学性质。结果表明,化合物c的紫外吸收波长、荧光发射波长、热稳定性、极化率均最大;化合物a—c均存在分子内电荷转移现象,非线性吸收均为双光子吸收,化合物a、b还有五阶非线性吸收,它们均表现出超快非线性光学响应,可作非线性光学研究备选材料。