应用化学
2024, 41 (
):
437-444.
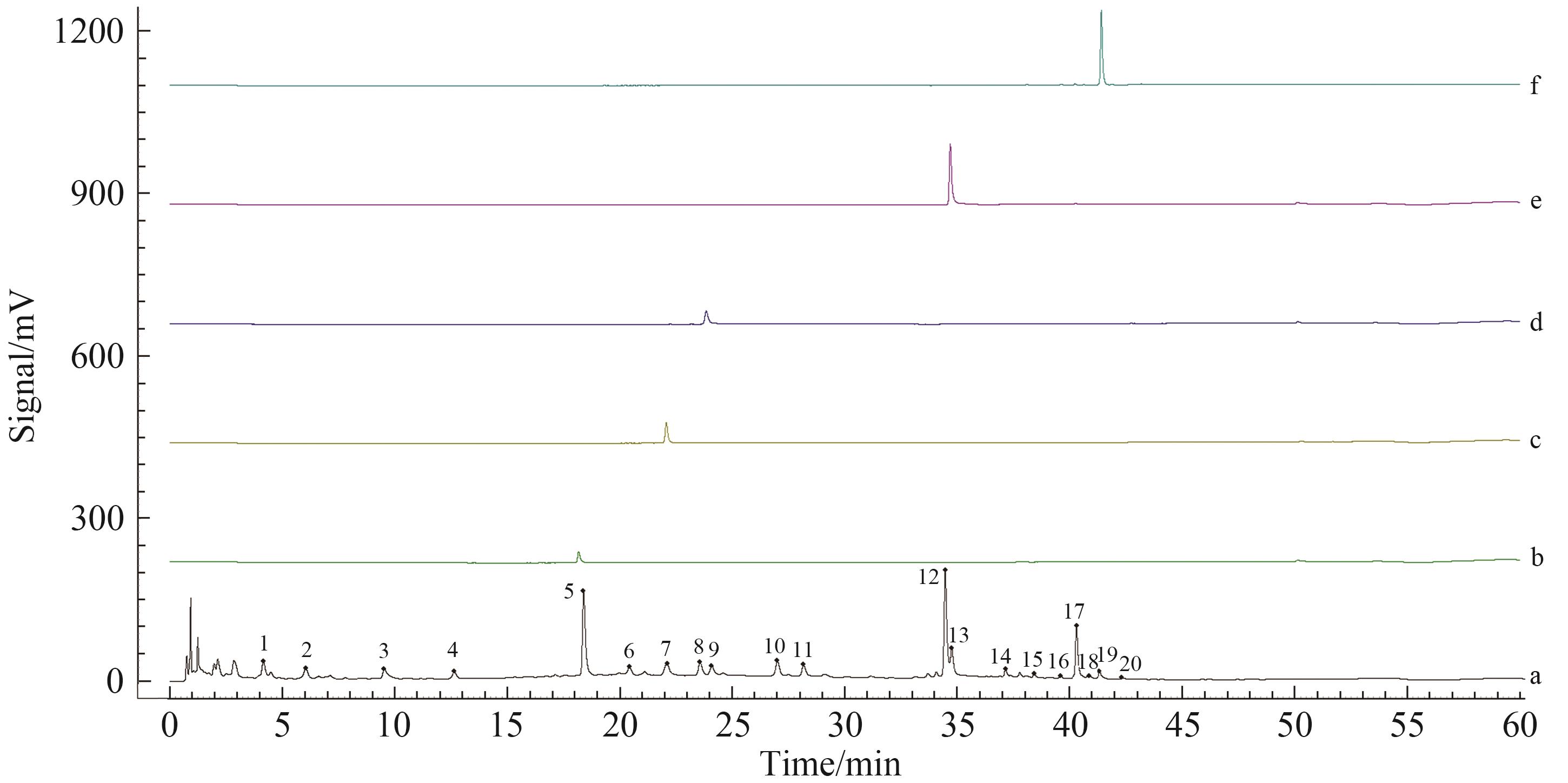
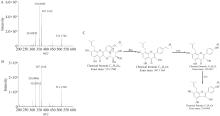
建立藤黄健骨丸高效液相色谱(HPLC)指纹图谱,完善产品质量标准。采用Waters Symmetry C18(150 mm×2.1 mm,3.5 μm)色谱柱; 流速为0.4 mL/min; 进样量为2 μL; 紫外检测波长为260 nm; 在柱温30 ℃下以0.1%甲酸水(A)和0.1%甲酸乙腈(B)为流动相进行梯度洗脱。利用“中药色谱指纹图谱相似度评价系统(2012版本)”建立藤黄健骨丸指纹图谱,并分析相似度。采用液质联用技术(UPLC-Q TOF-MSE),根据化合物串联质谱信息结合对照品进行了共有峰结构鉴定。共确定了10批藤黄健骨丸指纹图谱中20个共有峰,相似度均在0.95以上。基于对照品和串联质谱信息指认了共有峰中13种成分的结构,并进行了峰归属。对10批藤黄健骨丸指纹图谱共有峰的相对保留时间和相对峰面积进行了计算,结果显示各批次样品共有峰的相对保留时间和相对峰面积均比较稳定,表明它们所对应的各成分含量较为均一。采用该指纹图谱对中间体进行了相似度评价,发现中间体与成品相关性良好。该方法精密度、重复性和稳定性好,不仅可用于藤黄健骨丸成品的质量控制,还可对中间体进行检测,用于生产过程的控制。

{kind=link}