应用化学
2023, 40 (
):
1054-1060.
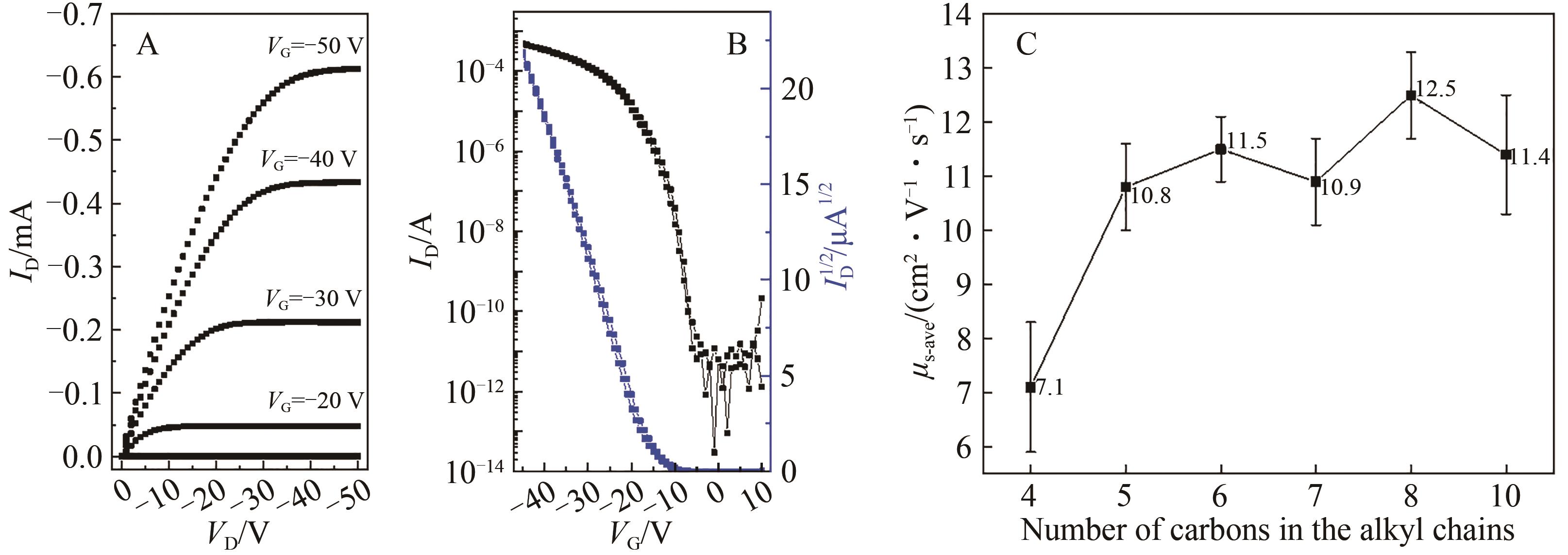
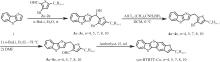
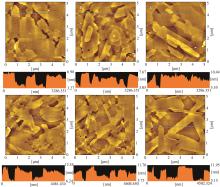
含噻吩并苯类分子是一类代表性的高迁移率有机半导体材料,以其为共轭骨架构建的不对称分子在薄膜中倾向于形成双层排列结构,并以二维层状方式生长,有利于实现高迁移率。烷基取代基的长度会对有机半导体材料的堆积形貌产生影响。本文设计合成了不同长度烷基链取代的噻吩并[4,5-b][1]苯并噻吩并[3,2-b][1]苯并噻吩(syn-BTBTT-Cn,n=4,5,6,7,8,10),系统研究了烷基链长度对化合物热稳定性、能级、载流子传输能力、堆积结构和薄膜形貌等方面的影响。结果表明,所有化合物均不具备液晶性,热稳定性良好。在所制备的蒸镀薄膜中所有分子均形成双层堆积结构,共轭核在层内形成鱼骨架堆积,烷基链长度会影响薄膜的有序度和堆积的紧密程度。基于该类材料制备的有机薄膜晶体管(OTFT)器件的迁移率都超过7.0 cm2/(V·s),其中syn-BTBTT-C8分子的迁移率最高可达13.8 cm2/(V·s),平均值为12.5 cm2/(V·s)。

{kind=link}