应用化学 ›› 2022, Vol. 39 ›› Issue (12): 1803-1817.DOI: 10.19894/j.issn.1000-0518.220053
张万年1, 于芳2,3, 赵杉林1,3, 张志强1( ), 何宇鹏1,2,3()
), 何宇鹏1,2,3()
收稿日期:2022-03-01
接受日期:2022-08-03
出版日期:2022-12-01
发布日期:2022-12-13
通讯作者:
张志强,何宇鹏
基金资助:
Wan-Nian ZHANG1, Fang YU2,3, Shan-Lin ZHAO1,3, Zhi-Qiang ZHANG1(), Yu-Peng HE1,2,3()
Received:2022-03-01
Accepted:2022-08-03
Published:2022-12-01
Online:2022-12-13
Contact:
Zhi-Qiang ZHANG,Yu-Peng HE
About author:heyp_nbi@dlut.edu.cnSupported by:摘要:
近年来,使用分子动力学(Molecular Dynamics, MD)模拟和汉森溶解度参数等计算方法,研究小分子凝胶行为备受关注。分子动力学模拟是一种基于经典力学的计算方法,用于理解小分子凝胶过程的首选技术之一,通过分子动力学模拟可以更精准地分析小分子凝胶的凝胶化趋势或组装行为,动态图形化地展现凝胶组装过程,有效揭示小分子凝胶因子结构与凝胶行为之间的关系,定量分析凝胶组装中的氢键、π-π堆积、范德华作用、离子键作用和疏溶剂作用等非共价键作用。通过对已知的凝胶/非凝胶分子进行分子动力学模拟,提取模拟数据中与凝胶行为相关的参数,并通过拟合Pearson相关系数衡量线性相关关系,最终实现预测某一类小分子是否可以凝胶化的目的。另一方面,根据汉森溶解度参数(Hansen Solubility Parameters, HSPs)发展形成的经验模型最具有代表性,该模型由分子之间的色散作用能量(δd)、极性作用能量(δp)和氢键能量(δh)确定三维空间(即汉森空间)的坐标点,根据该坐标点所在的范围可以确定有机小分子在特定的溶剂是否能形成凝胶。本文就近几年有机小分子凝胶领域分子动力学模拟和汉森溶解度参数中的一些工作进行综述,对凝胶的组装行为、非共价键作用对凝胶能力的调控和预测等方面作出评论。
中图分类号:
张万年, 于芳, 赵杉林, 张志强, 何宇鹏. 小分子凝胶的分子动力学模拟和汉森溶解度参数研究进展[J]. 应用化学, 2022, 39(12): 1803-1817.
Wan-Nian ZHANG, Fang YU, Shan-Lin ZHAO, Zhi-Qiang ZHANG, Yu-Peng HE. Progress in Molecular Dynamics and Hansen Solubility Parameters of Low Molecular Weight Gels[J]. Chinese Journal of Applied Chemistry, 2022, 39(12): 1803-1817.
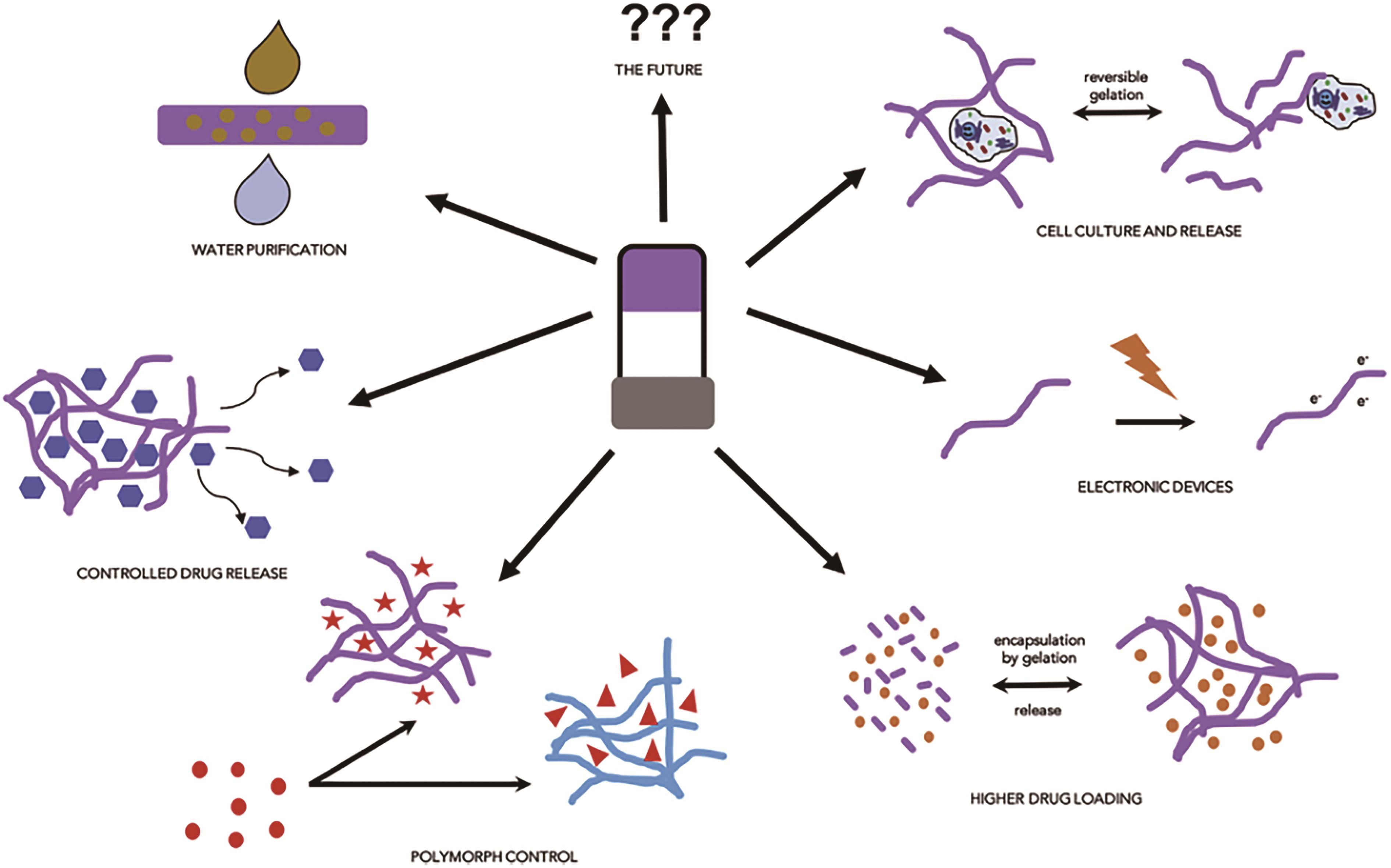
图1 小分子凝胶的应用与前景[18]
Fig.1 Application and future of LMWGs[18]
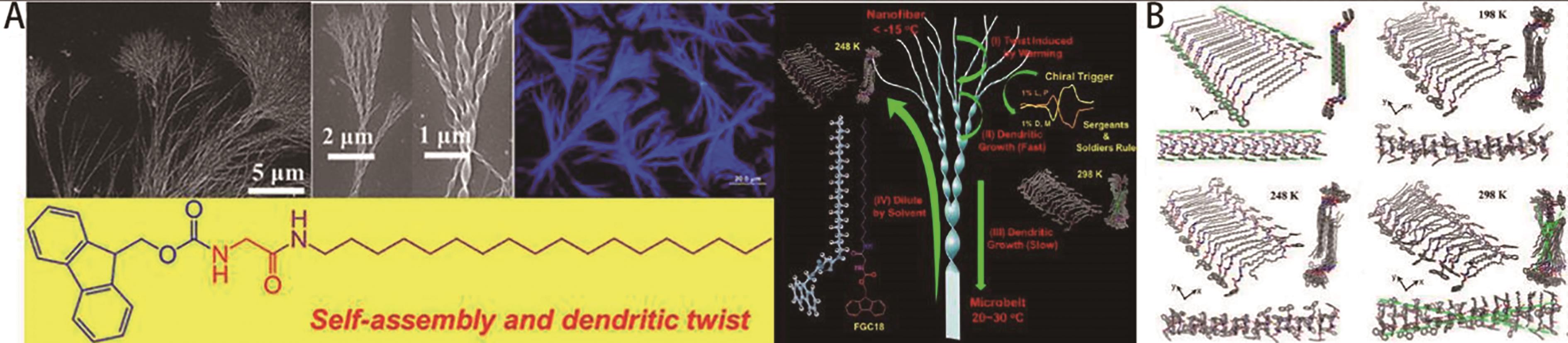
图2 (A)FGC18的分子结构和FGC18形成的分级枝晶扭曲结构示意图; (B)不同温度下,FGC18凝胶的分子动力学模拟快照[32]
Fig.2 (A) Molecular structure of FGC18 and the schematic illustration of hierarchical dendrite twist structure formed by FGC18; (B) Molecular dynamics simulation snapshots of FGC18 at different temperatures[32]

图3 (A) 纯脂质自组装的计算模型和机理; (B)手性脂质的自组装; (C)异手性脂质产生同手性纳米管的“诱导构象重排”机制[35]
Fig.3 (A) Calculation model and mechanism of the self-assembly of pure lipids; (B) Self-assembly of chiral lipids; (C) “Induced conformation rearrangement” mechanism of homochiral nanotube from heterochiral lipids[35]

图4 (A)12-HSAm聚合拓扑的模拟快照[36]; (B)双组分LMWG的分子动力学模拟的快照[37]
Fig.4 (A) Simulation snapshots of 12-HSAm aggregate topologies[36]; (B) Snapshots of MD simulation of two-component LMWGs[37]
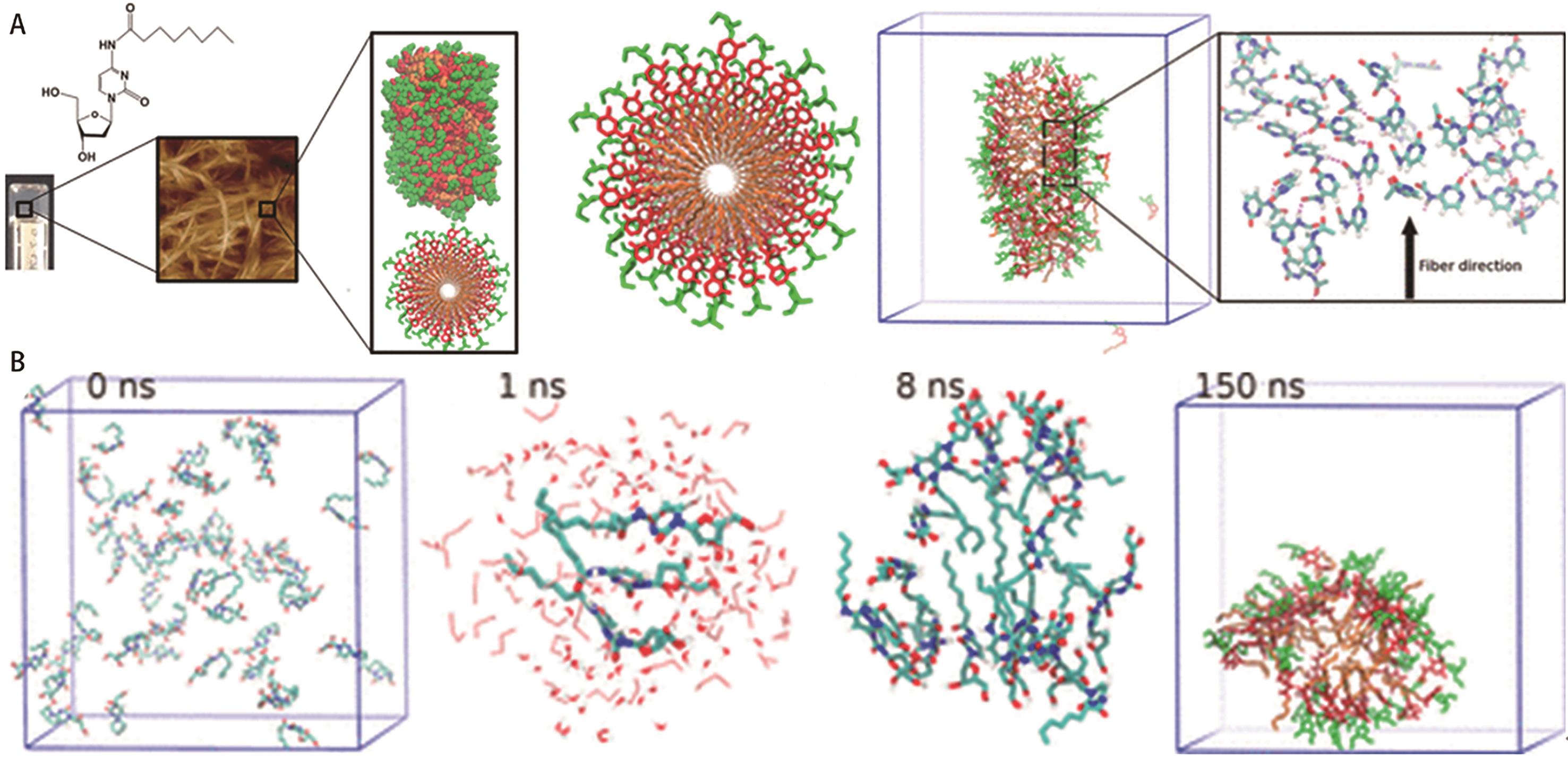
图5 (A)160个分子组成的团簇模型的凝胶分子在乙醇/水的MD模拟快照; (B)凝胶分子在乙醇/水(体积比20/80)体系中自组装的MD模拟快照[38]
Fig.5 (A) Snapshots from an MD simulation of 160 pre-ordered gelator molecules in ethanol/water; (B) Snapshots from an MD simulation of the self-assembly of 50 gelator molecules in ethanol/water (V/V, 20/80) system[38]

图6 (A)BTECM超分子柱状聚集体螺旋快照和平均构型的分子模型[43]; (B)甾醇有机凝胶的2个管模型在MD模拟过程的快照[45]; (C)2个小管之间的氢键、范德华作用和π-π堆积[45]
Fig.6 (A) Molecular models illustrating the average configurations in the supramolecular columnar aggregates of BTECM[43]; (B) Snapshots of two tubules of sitosterol-oryzanol and sitosterol-CHEMS at the beginning or at the end of the course of the MD simulation[45]; (C) Hydrogen bonds, vdW interactions and π-π contacts between the ferulic acid groups of oryzanol on the interface of two tubules[45]
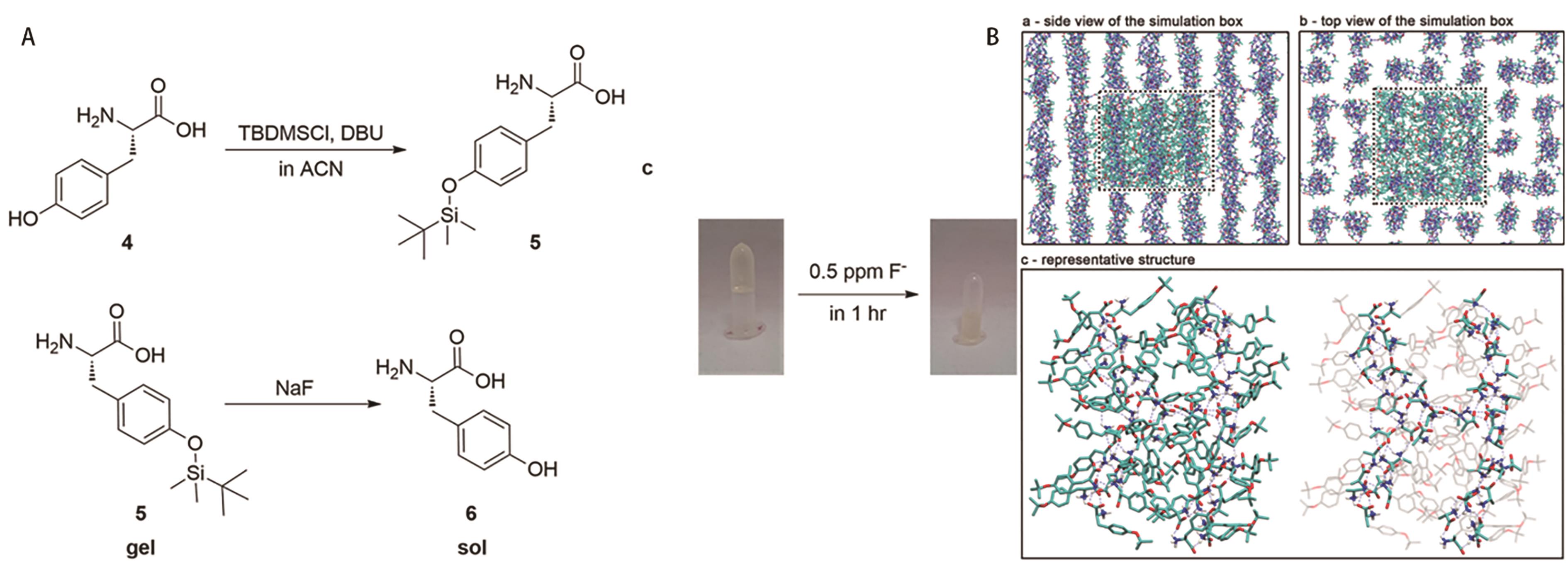
图7 (A)凝胶因子的合成与氟离子响应性示意图; (B)经1 μs MD模拟后凝胶自组装成纤维结构的快照[46]
Fig.7 (A) Schematic diagram of the synthesis of LMWGs and fluoride ion response; (B) Snapshot of the self-assembly of the gel into fibrous structures after 1 μs MD simulation[46]
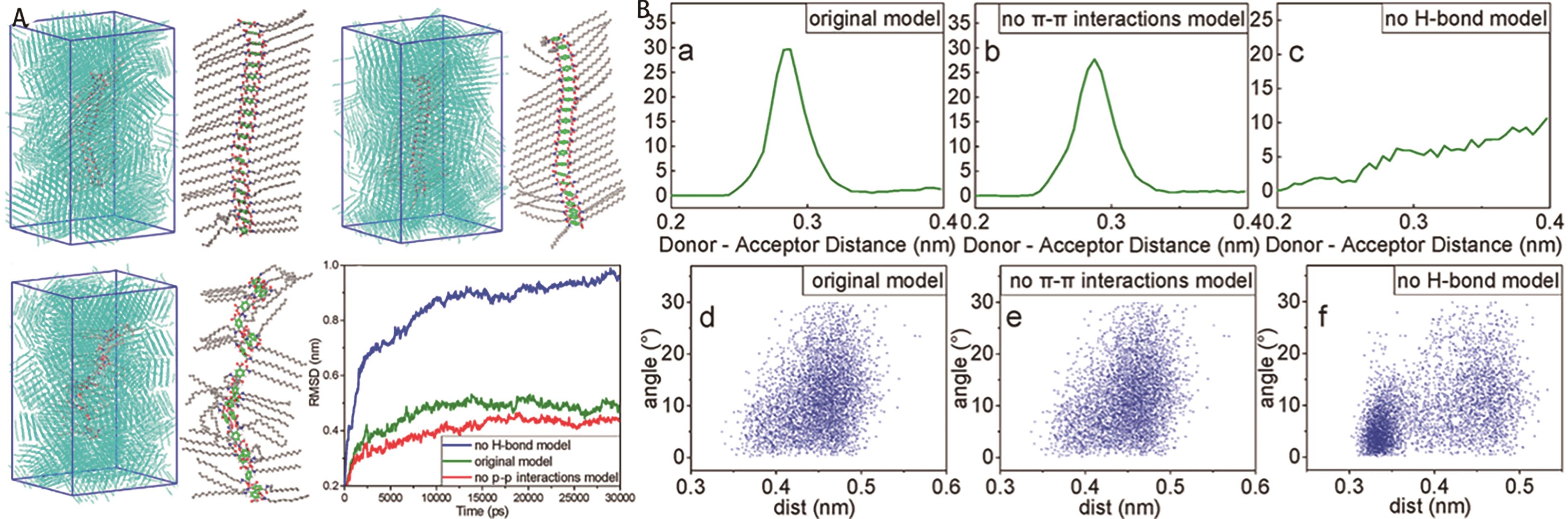
图8 (A)通过分子动力学模拟不同模型的凝胶自组装的平衡态; (B)在不同模型下氢键与π-π堆积的分布[47]
Fig.8 (A) The equilibrium states of gel self-assembly under different models by MD simulation; (B) The distribution of hydrogen bonds and π-π stacking under different models[47]
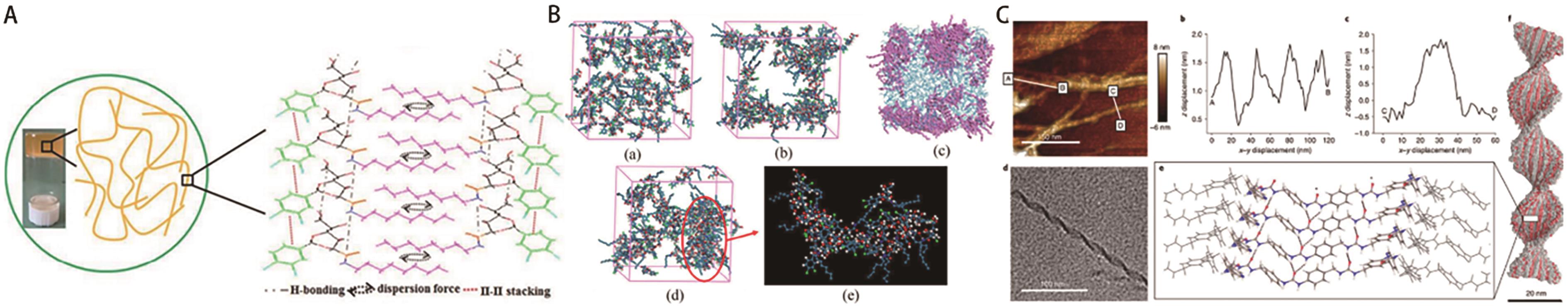
图9 (A)500SN凝胶自组装示意图;(B)500SN凝胶模拟结构的快照[52];(C)模拟形成的具有螺距和带厚度的稳定螺旋体[53]
Fig.9 (A) Schematic of self-assembly of the 500SN gel; (B) Snapshots of simulation structures of the 500SN gel[52]; (C) The simulations indicate that four layers are required to produce a stable helicoid with the observed pitch and ribbon thickness[53]
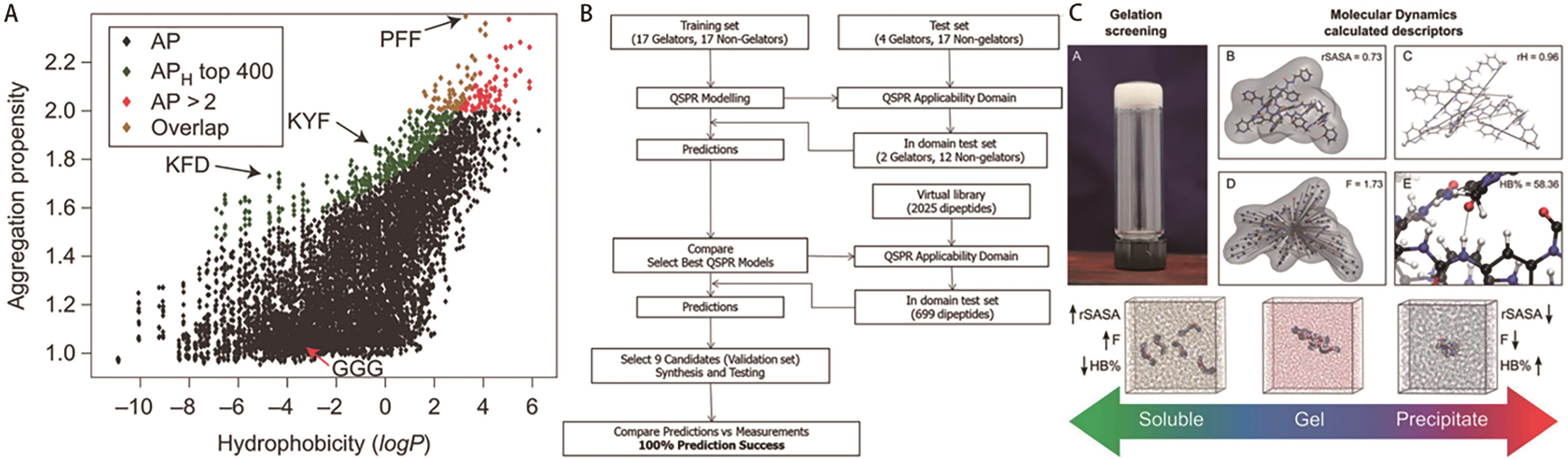
图10 (A)聚集倾向(APH)模型[27]; (B)整体QSPR建模、综合和测试工作流程[55]; (C)用描述符rSASA、F和HB%预测凝胶状态总体趋势[56]
Fig.10 (A) Aggregation propensity (APH)[27]; (B) Overall QSPR modelling, synthesis and testing workflow[55]; (C) Prediction of general trends in gel state with descriptors rSASA, F and HB%[56]

图11 Steed提出纤维凝胶的分级自组装阶段的模型[57]
Fig.11 Stages in the hierarchical self-assembly of fibrous gels according to the model proposed in steed study[57]
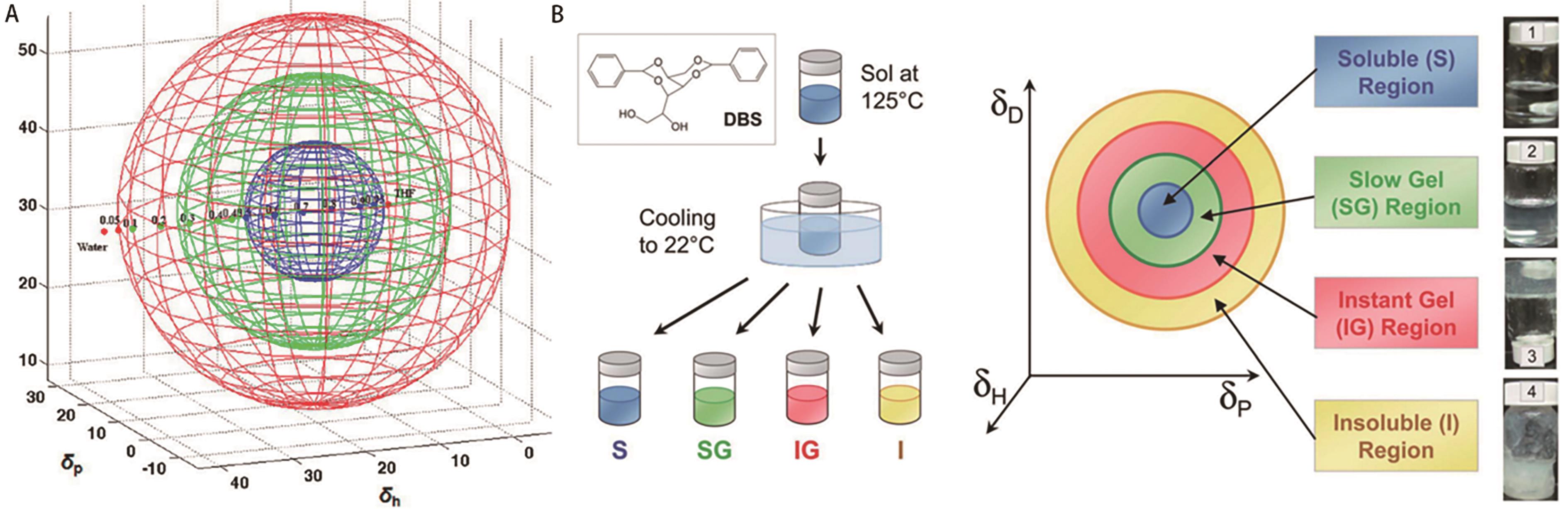
图12 (A)用汉森空间的球体表示2.0% LMWGs在液体混合物中的溶解度数据:蓝色(可溶)、绿色(凝胶)和红色(不溶)[62]; (B)实验装置示意图(左)和三维汉森空间中的相应结果(右)[63]
Fig.12 (A) Solubility data for 2.0% LMWGs in liquid mixtures represented in Hansen space with spheres/shells: blue (soluble), green (gel), and red (insoluble) [62]; (B) Schematic of the experimental setup (left) and the corresponding results in 3D Hansen space (right)[63]
| 1 | TERECH P, WEISS R G. Low molecular mass gelators of organic liquids and the properties of their gels[J]. Chem Rev, 1997, 97(8): 3133-3160. |
| 2 | LI Z, CAO J, LI H, et al. Self-assembled drug delivery system based on low-molecular-weight bis-amide organogelator: synthesis, properties and in vivo evaluation[J]. Drug Deliv, 2016, 23(8): 3168-3178. |
| 3 | LI J, GENG L, WANG G, et al. Self-healable gels for use in wearable devices[J]. Chem Mater, 2017, 29(21): 8932-8952. |
| 4 | 唐立宗, 张琳, 董云生, 等. 响应性水凝胶及其在生物医药领域应用研究进展[J]. 应用化学, 2021, 38(7): 743-753. |
| TANG L Z, ZHANG L, DONG Y S, et al. Research progress on responsive hydrogels and their applications in biomedicines[J]. Chinese J Appl Chem, 2021, 38(7): 743-753. | |
| 5 | LIM J Y C, GOH S S, LIOW S S, et al. Molecular gel sorbent materials for environmental remediation and wastewater treatment[J]. J Mater Chem A, 2019, 7(32): 18759-18791. |
| 6 | TRAUSEL F, VERSLUIS F, MAITY C, et al. Catalysis of supramolecular hydrogelation[J]. Acc Chem Res, 2016, 49(7): 1440-1447. |
| 7 | SHANG C, WANG G, HE M, et al. A high performance fluorescent arylamine sensor toward lung cancer sniffing[J]. Sens Actuators B: Chem, 2017, 241: 1316-1323. |
| 8 | TANAKA A, FUKUOKA Y, MORIMOTO Y, et al. Cancer cell death induced by the intracellular self-assembly of an enzyme-responsive supramolecular gelator[J]. J Am Chem Soc, 2015, 137(2): 770-775. |
| 9 | LIU X, FEI J, WANG A, et al. Transformation of dipeptide-based organogels into chiral crystals by cryogenic treatment[J]. Angew Chem Int Ed, 2017, 56(10): 2660-2663. |
| 10 | CHIVERS P R A, SMITH D K. Shaping and structuring supramolecular gels[J]. Nat Rev Mater, 2019, 4(7): 463-478. |
| 11 | DRAPER E R, ADAMS D J. How should multicomponent supramolecular gels be characterised?[J]. Chem Soc Rev, 2018, 47(10): 3395-3405. |
| 12 | BACANI R. Gel characterization: from molecules to nanostructure to macroproperties[M]. Nano Design for Smart Gels, 2019: 141-206. |
| 13 | WU S, ZHANG Q, DENG Y, et al. Assembly pattern of supramolecular hydrogel induced by lower critical solution temperature behavior of low-molecular-weight gelator[J]. J Am Chem Soc, 2020, 142(1): 448-455. |
| 14 | DAMA M, BERGER S. Study of an organogelator by diffusion-ordered NMR spectroscopy[J]. J Phys Chem B, 2013, 117(18): 5788-5791. |
| 15 | ADAMS D J, MORRIS K, CHEN L, et al. The delicate balance between gelation and crystallisation: structural and computational investigations[J]. Soft Matter, 2010, 6(17): 4144-4156. |
| 16 | CICCHI S, GHINI G, LASCIALFARI L, et al. Self-sorting chiral organogels from a long chain carbamate of 1-benzyl-pyrrolidine-3,4-diol[J]. Soft Matter, 2010, 6(8): 1655-1661. |
| 17 | WEISS R G. Controlling variables in molecular gel science: how can we improve the state of the art?[J]. Gels, 2018, 4(2): 25. |
| 18 | DRAPER E R, ADAMS D J. Low-molecular-weight gels: the state of the art[J]. Chem, 2017, 3(3): 390-410. |
| 19 | OTOISHI Y, SUGITA Y. Metabolite-protein interactions in bacterial cytoplasm: all-atomic cytoplasm model and theoretical study by large-scale molecular dynamics calculations[J]. Ensemble, 2017, 19: 75-80. |
| 20 | BABU S S, PRAVEEN V K, AJAYAGHOSH A. Functional pi-gelators and their applications[J]. Chem Rev, 2014, 114(4): 1973-2129. |
| 21 | ZHANG W, WANG K, WANG C, et al. Enhanced oil recovery: QM/MM based descriptors for anionic surfactant salt-resistance[J]. Colloid Surface A, 2022, 641: 128422. |
| 22 | 刘旭, 李杨可欣, 杜黎, 等 水凝胶的制备及仿生设计在能源领域应用的研究进展[J]. 应用化学, 2022, 39(1): 35-54. |
| LIU X, LI Y K X, DU L, et al. Bio⁃inspired hydrogels: synthesis, bionic design and applications in the field of energy storage and conversion[J]. Chinese J Appl Chem, 2022, 39(1): 35-54. | |
| 23 | 李胜男, 付俊. 水凝胶仿生柔性电子学[J]. 应用化学, 2022, 39(1): 55-73. |
| LI S N, FU J. Biomimetic flexible hydrogel electronics[J]. Chinese J Appl Chem, 2022, 39(1): 55-73. | |
| 24 | SULIMOV A V, KUTOV D C, KATKOVA E V, et al. New generation of docking programs: supercomputer validation of force fields and quantum-chemical methods for docking[J]. J Mol Graph Model, 2017, 78: 139-147. |
| 25 | YAN Y, TAO H, HE J, et al. The HDOCK server for integrated protein-protein docking[J]. Nat Protoc, 2020, 15(5): 1829-52. |
| 26 | JUMPER J, EVANS R, PRITZEL A, et al. Highly accurate protein structure prediction with AlphaFold[J]. Nature, 2021, 596(7873): 583-589. |
| 27 | FREDERIX P W, SCOTT G G, ABUL-HAIJA Y M, et al. Exploring the sequence space for (tri-)peptide self-assembly to design and discover new hydrogels[J]. Nat Chem, 2015, 7(1): 30-37. |
| 28 | KULKARNI C, KOREVAAR P A, BEJAGAM K K, et al. Solvent clathrate driven dynamic stereomutation of a supramolecular polymer with molecular pockets[J]. J Am Chem Soc, 2017, 139(39): 13867-13875. |
| 29 | MATHESON A, DALKAS G, MEARS R, et al. Stable emulsions of droplets in a solid edible organogel matrix[J]. Soft Matter, 2018, 14(11): 2044-2051. |
| 30 | XING P, CHEN H, XIANG H, et al. Selective coassembly of aromatic amino acids to fabricate hydrogels with light irradiation-induced emission for fluorescent imprint[J]. Adv Mater, 2018, 30(5): 1705633. |
| 31 | CHRISTOFF-TEMPESTA T, LEW A J, ORTONY J H. Beyond covalent crosslinks: applications of supramolecular gels[J]. Gels, 2018, 4(2): 40. |
| 32 | CAO H, YUAN Q, ZHU X, et al. Hierarchical self-assembly of achiral amino acid derivatives into dendritic chiral nanotwists[J]. Langmuir, 2012, 28(43): 15410-15417. |
| 33 | GREEN MARK M, PETERSON NORMAN C, SATO T, et al. A helical polymer with a cooperative response to chiral information[J]. Science, 1995, 268(5219): 1860-1866. |
| 34 | LANGEVELD-VOSS B M W, WATERVAL R J M, JANSSEN R A J, et al. Principles of “majority rules” and “sergeants and soldiers” applied to the aggregation of optically active polythiophenes: evidence for a multichain phenomenon[J]. Macromolecules, 1999, 32(1): 227-230. |
| 35 | ZHU X, JIANG Y, YANG D, et al. Homochiral nanotubes from heterochiral lipid mixtures: a shorter alkyl chain dominated chiral self-assembly[J]. Chem Sci, 2019, 10(13): 3873-3880. |
| 36 | HUDA M M, RAI N. Probing early-stage aggregation of low molecular weight gelator in an organic solvent[J]. J Phys Chem B, 2020, 124(11): 2277-2288. |
| 37 | SU T, HONG K H, ZHANG W, et al. Scaleable two-component gelator from phthalic acid derivatives and primary alkyl amines: acid-base interaction in the cooperative assembly[J]. Soft Matter, 2017, 13(22): 4066-4073. |
| 38 | ANGELEROU M G F, FREDERIX P, WALLACE M, et al. Supramolecular nucleoside-based gel: molecular dynamics simulation and characterization of its nanoarchitecture and self-assembly mechanism[J]. Langmuir, 2018, 34(23): 6912-6921. |
| 39 | DASTIDAR P. Supramolecular gelling agents: can they be designed?[J]. Chem Soc Rev, 2008, 37(12): 2699-2715. |
| 40 | CHUANG P H, TSENG Y H, GU Q, et al. Phase behavior and electron transfer properties of ferrocenyl cholesteryl N-formanidoformamide gelator: a computational study[J]. Colloid Polym Sci, 2015, 293(7): 2113-2119. |
| 41 | KULKARNI C, MEIJER E W, PALMANS A R A. Cooperativity scale: a structure-mechanism correlation in the self-assembly of benzene-1,3,5-tricarboxamides[J]. Acc Chem Res, 2017, 50(8): 1928-1936. |
| 42 | ALEGRE-REQUENA J V, SALDIAS C, INOSTROZA-RIVERA R, et al. Understanding hydrogelation processes through molecular dynamics[J]. J Mater Chem B, 2019, 7(10): 1652-1673. |
| 43 | SHEN Z, JIANG Y, WANG T, et al. Symmetry breaking in the supramolecular gels of an achiral gelator exclusively driven by pi-pi stacking[J]. J Am Chem Soc, 2015, 137(51): 16109-16115. |
| 44 | SHEN Z, WANG T, LIU M. Macroscopic chirality of supramolecular gels formed from achiral tris(ethyl cinnamate) benzene-1, 3, 5-tricarboxamides[J]. Angew Chem Int Ed, 2014, 53(49): 13424-13428. |
| 45 | DALKAS G, MATHESON A B, VASS H, et al. Molecular interactions behind the self-assembly and microstructure of mixed sterol organogels[J]. Langmuir, 2018, 34(29): 8629-8638. |
| 46 | AYKENT G, ZEYTUN C, MARION A, et al. Simple tyrosine derivatives act as low molecular weight organogelators[J]. Sci Rep, 2019, 9(1): 4893. |
| 47 | ZHANG W, ZHANG Z, ZHAO S, et al. Pyromellitic-based low molecular weight gelators and computational studies of intermolecular interactions: a potential additive for lubricant[J]. Langmuir, 2021, 37(9): 2954-2962. |
| 48 | SINGHA N, SRIVASTAVA A, PRAMANIK B, et al. Unusual confinement properties of a water insoluble small peptide hydrogel[J]. Chem Sci, 2019, 10(23): 5920-5928. |
| 49 | SHI L, HAN Q. Molecular dynamics study of the influence of solvents on the structure and mechanical properties of poly(vinyl alcohol) gels[J]. J Mol Model, 2018, 24(11): 325. |
| 50 | VYAS S, KHAMBETE M, GUDHKA R, et al. Network topology of LMWG cross-linked xyloglucan hydrogels for embedding hydrophobic nanodroplets: mechanistic insight and molecular dynamics[J]. Drug Deliv Transl Res, 2020, 10(4): 1076-1084. |
| 51 | GUILBAUD-CHEREAU C, DINESH B, SCHURHAMMER R, et al. Protected amino acid-based hydrogels incorporating carbon nanomaterials for near-infrared irradiation-triggered drug release[J]. ACS Appl Mater Interfaces, 2019, 11(14): 13147-13157. |
| 52 | WANG Y, YU Q, BAI Y, et al. Self-constraint gel lubricants with high phase transition temperature[J]. ACS Sustain Chem Eng, 2018, 6(11): 15801-15810. |
| 53 | JONES C D, SIMMONS H T D, HORNER K E, et al. Braiding, branching and chiral amplification of nanofibres in supramolecular gels[J]. Nat Chem, 2019, 11(4): 375-381. |
| 54 | BAI Y, YU Q, ZHANG J, et al. Soft-nanocomposite lubricants of supramolecular gel with carbon nanotubes[J]. J Mater Chem A, 2019, 7(13): 7654-7663. |
| 55 | GUPTA J K, ADAMS D J, BERRY N G. Will it gel? successful computational prediction of peptide gelators using physicochemical properties and molecular fingerprints[J]. Chem Sci, 2016, 7(7): 4713-4719. |
| 56 | VAN LOMMEL R, ZHAO J, DE BORGGRAEVE W M, et al. Molecular dynamics based descriptors for predicting supramolecular gelation[J]. Chem Sci, 2020, 11(16): 4226-4238. |
| 57 | JONES C D, KENNEDY S R, WALKER M, et al. Scrolling of supramolecular lamellae in the hierarchical self-assembly of fibrous gels[J]. Chem, 2017, 3(4): 603-628. |
| 58 | HANSEN C. Hansen solubility parameters: a user's handbook, Second Edition[M]. USA: CRC Press, 2012. |
| 59 | 刘建强, 马婧. 用汉森溶解度参数评估功能化石墨烯在溶剂中的分散性[J]. 过程工程学报, 2019, 19(1): 189-194. |
| LIU J Q, MA J. Evaluation of dispersion of functionalized graphene in solvents by Hansen solubility parameters[J]. Chinese J Process Eng, 2019, 19(1): 189-194. | |
| 60 | RAYNAL M, BOUTEILLER L. Organogel formation rationalized by Hansen solubility parameters[J]. Chem Commun, 2011, 47(29): 8271-8273. |
| 61 | ROSA NUNES D, RAYNAL M, ISARE B, et al. Organogel formation rationalized by Hansen solubility parameters: improved methodology[J]. Soft Matter, 2018, 14(23): 4805-4809. |
| 62 | YAN N, XU Z, DIEHN K K, et al. How do liquid mixtures solubilize insoluble gelators? self-assembly properties of pyrenyl-linker-glucono gelators in tetrahydrofuran-water mixtures[J]. J Am Chem Soc, 2013, 135(24): 8989-8999. |
| 63 | DIEHN K K, OH H, HASHEMIPOUR R, et al. Insights into organogelation and its kinetics from Hansen solubility parameters. Toward a priori predictions of molecular gelation[J]. Soft Matter, 2014, 10(15): 2632-2640. |
| 64 | LAN Y, CORRADINI M G, WEISS R G, et al. To gel or not to gel: correlating molecular gelation with solvent parameters[J]. Chem Soc Rev, 2015, 44(17): 6035-6058. |
| 65 | RIWAR L J, TRAPP N, KUHN B, et al. Substituent effects in parallel-displaced pi-pi stacking interactions: distance matters[J]. Angew Chem Int Ed, 2017, 56(37): 11252-11257. |
| 66 | EMAMIAN S, LU T, KRUSE H, et al. Exploring nature and predicting strength of hydrogen bonds: a correlation analysis between atoms-in-molecules descriptors, binding energies, and energy components of symmetry-adapted perturbation theory[J]. J Comput Chem, 2019, 40(32): 2868-2881. |
| 67 | LU T, CHEN Q. Van der Waals potential: an important complement to molecular electrostatic potential in studying intermolecular interactions[J]. J Mol Model, 2020, 26(11): 315. |
| 68 | WEISS R G. The past, present, and future of molecular gels. what is the status of the field, and where is it going?[J]. J Am Chem Soc, 2014, 136(21): 7519-7530. |
| [1] | 邓培渊, 袁伟, 李长看, 陈龙欣, 杨莹莹. 防腐剂苯甲酸与人血清白蛋白相互作用[J]. 应用化学, 2021, 38(8): 1014-1021. |
| [2] | 郑丹丹,张颖. 环保型阻垢剂对水垢的抑制及其相互作用模拟[J]. 应用化学, 2019, 36(11): 1308-1316. |
| [3] | 杨科成, 崔凤超, 李云琦. 核糖核酸酶Sa表面水和尿素分子的分布和动力学行为的分子动力学模拟[J]. 应用化学, 2018, 35(10): 1243-1248. |
| [4] | 杨登峰, 刘清芝, 李红曼, 高从堦. 端基改性碳纳米管膜分离Li+/Mg2+的分子动力学模拟[J]. 应用化学, 2014, 31(11): 1345-1351. |
| [5] | 杜满泉, 史林启, 安英丽, 李湛勇, 何炳林. 聚苯乙烯-丙烯酸嵌段共聚物在Ca2+或乙二胺存在下的自组装行为[J]. 应用化学, 2001, 18(11): 915-917. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||