{kind=link}
22-降-豆甾-22-含氮化合物的合成及抗肿瘤活性
[杨雷a , 范良华b , 赵丹丹a , 戚斌斌a , 甘春芳a , 崔建国a , 黄燕敏a, *  ]
]
]
 |


从豆甾醇出发,通过3-位羟基及5-位双键保护, 22-位双键臭氧化,得到3-乙酰氧基胆甾-5-烯-22-醛(4)。 化合物4进一步与氨基脲、氨基硫脲缩合得到22-降豆甾-22-含氮衍生物,合成物结构经NMR、IR及MS等技术手段进行了表征。 并采用6种不同的肿瘤细胞,利用MTT法对合成物进行体外抗肿瘤活性研究,结果表明,具有22-缩氨硫脲基的化合物8具有很好的体外抗肿瘤活性,其对MGC 7901、Hela、SMMC 7404及CNE-2细胞的IC50值分别为10.5、11.6、8.7及11.9。
Using stigmasterol as a starting material, 3 β-acetoxycholest-5-ene-22-al was obtained by the protection of 3-hydroxyl and 5-double bond prior to the ozonolysis of 22-ene. Some 22-abeo-stigmast-22-nitrogen-containing compounds were synthesized by the condensation of aldehydes with semicarbazides and thiosemicarbazides. The structure of compounds were confirmed with NMR, IR and MS. Their antiproliferative activities against six kinds of cancer cells were evaluated with MTT method. Compound 8 with the 22-thiosemicarbazone group exhibits significant inhibitory activity on MGC 7901, Hela, SMMC 7404 and CNE-2 cells with the IC50 values of 10.5, 11.6, 8.7 and 11.9 μmol/L, respectively.
甾体含氮化合物具有显著的生理活性。 近年来, 甾体药物在医疗领域中的应用范围不断扩大[1, 2, 3], 甾体腙类化合物及其衍生物由于具有较好的抗菌、抗HIV、抗肿瘤等多种生理活性从而被广泛研究, 并得到迅速的发展[4, 5, 6, 7]。 在前期研究中, 我们合成了一系列的甾体肟类化合物及甾体内酰胺化合物[8, 9, 10, 11], 得到了一些在体内及体外均具有很好生理活性的甾体含氮化合物[12]。 同时在对这些甾体化合物的合成及抗肿瘤活性筛选中我们发现, 具有胆甾醇结构支链的甾体肟类化合物其细胞毒性远比具有豆甾醇或谷甾醇支链结构的甾体肟类化合物的细胞毒性小, 为了进一步探索甾体化合物中支链的结构对化合物母体细胞毒性的影响, 本文合成了一些具有胆甾醇甾核结构及不同支链的甾体化合物(Scheme 1所示), 探索了它们的抗肿瘤细胞生长增殖活性。
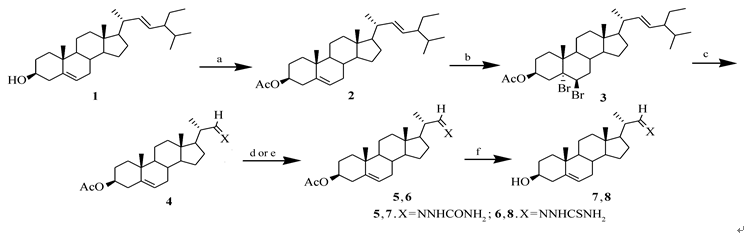 | Scheme 1 Synthesis of 22-abeo-stigmast-22-nitrogen-containing compoundsReagents and conditions:a.Ac2O/Pyridine/r.t., 24 h; b.Br2, PhI, hexane; c.1)O3/CH2Cl2, -78 ℃; 2)Zn/AcOH, 12 h; d.NH2NHCONH2/AcOH, EtOH (for compound 5); e.NH2NHCSNH2/AcOH, EtOH(for compound 6); f.m(KOH)∶ m(MeOH)=5∶ 95 |
合成所用试剂均为分析纯, 溶剂按常规方法进行处理, 干燥。 红外光谱测定采用Nicolet FT 360型红外光谱仪(美国赛默飞世尔公司), KBr压片; 1H 和13C核磁共振谱测定采用Bruker AV-300(300 MHz)型超导核磁共振仪(德国布鲁克公司), δ 表示化学位移, 偶合常数J用Hz表示, TMS为内标; 熔点测定采用X-6型显微熔点测定仪(上海仪电物理光学仪器有限公司制造), 温度计未经校正, TLC板为GF254硅胶板, 柱层析所用的硅胶为48~75 μ m的硅胶。
化合物4的合成参照参考文献[13]中的合成方法进行。
1.2.1 22-降-3β -乙酰氧基胆甾-5-烯-22-缩氨脲(5)的合成 在装有球形冷凝管的反应瓶中将75 mg(0.2 mmol)3β -乙酰氧基胆甾-5-烯-22-醛溶于15 mL 95%乙醇, 加入35 mg(0.25 mmol)三水乙酸钠, 搅拌10 min, 溶液呈透明。 保持反应温度在65 ℃, 一次性加入28 mg(0.25 mmol)盐酸氨基脲, 溶液呈黄色。 TLC监测反应(V(甲醇)∶ V(二氯甲烷)=1∶ 20), 反应1 h无原料点后停止搅拌。 减压蒸出大部分溶剂, 出现大量的淡黄色固体, 加入少量水, 分别用乙酸乙酯萃取3次(15 mL× 3), 合并有机层, 水洗、饱和食盐水洗, 然后用无水硫酸钠干燥。 减压蒸去溶剂得到淡黄色固体80 mg, 粗产品经柱层析分离(洗脱剂:V(甲醇)∶ V(二氯甲烷)=1∶ 20)得到69 mg淡黄色固体产物5, 产率80%, mp 213~215 ℃。 IR(KBr), σ /cm-1:3468, 3230, 2941, 2864, 1732, 1687, 1585, 1442, 1372, 1245, 1139, 1033; 1H NMR(300 MHz, CDCl3), δ :0.736(s, 3H, 18-CH3), 1.039(s, 3H, 19-CH3), 1.132(d, 3H, J=6.9 Hz, 21-CH3), 2.049(s, 3H, 3-OCOCH3), 2.39~2.28(m, 2H, C4— H), 4.67~4.56(m, 1H, C3— α H), 5.388(d, 1H, J=4.8 Hz, C6— H), 6.933(d, 1H, J=6.9 Hz, C22— H), 8.549(s, 1H, NH— ); 13C NMR(75 MHz, CDCl3), δ :170.6(3-COCH3), 149.9(— NHCONH2), 139.7(5-C), 139.6(22-C), 122.5(6-C), 73.9(3-C), 56.4(14-C), 53.9(17-C), 49.9(9-C), 42.6(13-C), 39.5(12-C), 38.1(4-C), 37.0(10-C), 36.6(1-C), 31.8(20-C, 7-C, 8-C), 27.7(2-C), 27.6(16-C), 24.4(15-C), 21.4(3-CH3CO— ), 21.0(11-C), 19.3(19-C), 17.7(21-C), 12.1(18-C); ESI-MS: 430.3(M+1)+。
1.2.2 22-降-3β -乙酰氧基胆甾-5-烯-22-缩氨硫脲(6)的合成 在连有球形冷凝管的反应瓶中将67 mg(0.18 mmol)化合物4溶于20 mL无水乙醇, 滴入冰醋酸调节溶液pH值为3~5。 搅拌反应, 保持反应温度在70 ℃。 完全溶解后, 一次性加入32 mg(0.36 mmol)氨基硫脲, 溶液呈淡黄色。 TLC跟踪反应(V(甲醇)∶ V(二氯甲烷)=1∶ 20), 反应1 h, 无原料点后停止搅拌。 减压蒸出大部分溶剂, 出现大量的淡黄色固体, 停止蒸馏, 加入少量水(2 mL), 分别用乙酸乙酯萃取3次(10 mL× 3), 合并有机层, 饱和碳酸氢钠洗1次, 水洗1次, 饱和食盐水洗1次, 然后用无水硫酸钠干燥。 蒸去溶剂得到淡黄色固体。 粗产品经柱层析分离(洗脱剂:V(甲醇)∶ V(二氯甲烷)=1∶ 20)得到62 mg淡黄色固体产物6, 产率89%, mp 204~210 ℃。 IR(KBr), σ /cm-1:3436, 3264, 2937, 2872, 1728, 1597, 1531, 1458, 1245, 1037, 612; 1H NMR(300 MHz, CDCl3), δ :0.735(s, 3H, 18-CH3), 1.038(s, 3H, 19-CH3), 1.150(d, 3H, J=6.0 Hz, 21-CH3), 2.050(s, 3H, COCH3), 2.40~2.30(m, 2H, C4— H), 4.68~4.54(m, 1H, C3— α H), 5.388(br s, 1H, C6— H), 6.275(s, 1H, — NH2), 7.06(br s, 1H, — NH2), 7.102(d, 1H, J=6.9 Hz, C22— H), 9.256(s, 1H, — NH— ); 13C NMR(75 MHz, CDCl3), δ :178.3( 
1.2.3 22-降-3β -羟基胆甾-5-烯-22-缩氨脲(7)的合成 40 mg化合物5溶于10 mL KOH/CH3OH(体积比5∶ 95)中, 在室温下搅拌反应, TLC跟踪反应(V(石油醚)∶ V(乙酸乙酯)=2∶ 1), 1 h后几乎不见原料点, 停止反应。 减压蒸去溶剂得白色固体, 加入10 mL冰水, 用乙酸乙酯萃取3次(10 mL× 3), 合并有机层后用水洗1次, 饱和食盐水洗1次, 无水硫酸钠干燥。 过滤, 减压除去溶剂得透明的油状物。 柱层析分离(洗脱剂:V(石油醚)∶ V(乙酸乙酯)=2∶ 1)得到30 mg白色固体化合物7 , 产率83%, mp 241~243 ℃。 IR(KBr), σ /cm-1:3477, 3276, 2929, 2864, 1695, 1585, 1446, 1372, 1135, 1057; 1H NMR(300 MHz, CDCl3), δ :0.744(s, 3H, 18-CH3), 1.032(s, 3H, 19-CH3), 1.140(d, 3H, J=6.0 Hz, 21-CH3), 2.39~2.25(m, 2H, C4— H), 3.60~3.50(m, 1H, C3— α H), 5.370(s, 1H, C6— H), 6.901(d, 1H, J=6.6 Hz, C22— H), 7.851(s, 1H, — NH— ); 13C NMR(75 MHz, CDCl3), δ :150.6(— NHCONH2), 140.8(5-C), 140.7(22-C), 121.0(6-C), 71.0(3-C), 56.5(14-C), 54.0(17-C), 50.3(9-C), 42.3(13-C), 41.6(4-C), 39.4(12-C), 39.3(1-C), 37.1(10-C), 36.3(20-C), 31.8(7-C), 31.6(8-C), 30.9(2-C), 27.1(16-C), 24.0(15-C), 20.7(11-C), 18.5(19-C), 16.7(21-C), 11.2(18-C); HREIMS:m/z 388.2946[M+H]+(计算值C23H38N3O2, 388.2964)。
1.2.4 22-降-3β -羟基胆甾-5-烯-22-缩氨硫脲(8)的合成 合成方法与化合物7的合成方法类似, 化合物8的产率89%, mp 216 ~ 217 ℃。 IR(KBr), σ /cm-1:3432, 3260, 2933, 2856, 1593, 1527, 1452, 1376, 1282, 1053; 1H NMR(300 MHz, CDCl3), δ :0.735(s, 3H, 18-CH3), 1.024(s, 3H, 19-CH3), 1.147(d, 3H, J=6.3 Hz, 21-CH3), 2.42~2.24(m, 2H, C4-H), 3.60~3.48(m, 1H, C3— α H), 5.362(br s, 1H, C6— H), 6.288(br s, 1H, — NH2), 7.05(br s, 1H, — NH2), 7.095(d, 1H, J=7.2 Hz, C22— H), 9.266(s, 1H, — NH— ); 13C NMR(75 MHz, CDCl3), δ :178.4( 
使用顺铂(Cisplatin)作为阳性对照, 采用MTT(methyl thiazolyl tetrazolium, MTT)法测试所合成的目标化合物对人体胃癌细胞株(SGC 7901), 人体宫颈癌细胞株(HeLa), 人体肝癌细胞株(Bel-7404), 人体肺癌细胞株(SPC-A), 人体鼻咽癌细胞株(CNE2)和人体喉癌细胞株(TU 686)的体外生长增殖抑制活性。
实验步骤如下:步骤1、首先将有机化合物溶解在DMSO中, 配制成10 g/L的DMSO溶液保存在冰箱; 步骤2、将对数生长期的肿瘤细胞分别以3× 104~4× 104个/mL 的密度接种于96孔板中, 每孔接种 200 μ L, 置于CO2培养箱中培养24 h; 步骤3、 按预设的浓度梯度(最高浓度设为100 mg/L)加入待测样品, 每一浓度梯度设3个平行孔, 同时设对照孔和空白孔, 对照孔和空白孔中加入最高浓度孔所需的等体积的DMSO(2 μ L); 步骤4、在二氧化碳培养箱中于37 ℃培养72 h后, 每孔加入20 μ L 的MTT(5 g/L), 然后在CO2培养箱中继续温育4 h。 抽取上清液, 然后加入200 μ L的DMSO, 在振动器上震荡10 min溶解沉淀, 随后用酶标仪测定光密度(OD)值。 通过下式求出一定浓度下样品对细胞的抑制率:
抑制率/%=[(对照OD-空白OD)-(给药OD-空白OD)]/(对照OD-空白OD)× 100
然后以抑制率对药物浓度作图, 求出每个样品的IC50值(50%抑制浓度)。 如果化合物在最高浓度没有抑制作用, 一般测定一次, 其它的每个样品测定3次, 计算平均值, 偏差大的需要进一步测定。
从豆甾醇出发, 对3-羟基通过乙酰化进行保护。 而5, 6-双键通过溴代进行保护, 在这里由于5, 6-双键是三取代双键, 而22, 23-双键是二取代双键, 因此5, 6-双键反应活性高于22, 23-双键, 优先发生溴代反应得到化合物3。 化合物3在低温进行臭氧化反应后, 采用锌粉/醋酸进行分解得到22-醛基, 同时甾核中的5, 6-双键发生去保护脱去溴原子得到化合物4。 化合物4分别与氨基脲及氨基硫脲反应得到化合物5和6, 化合物5和6在碱性条件下脱去3-乙酰基保护基最后得到化合物7和8。
化合物5的红外光谱中, 在3120 cm-1处存在氨基吸收峰。 从该化合物的1H NMR中可以看出, δ 8.549和5.750处出现— NH的化学位移, δ 6.922~6.945为双峰, 是 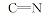
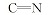
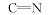
化合物6的红外光谱中, 在3154、3264和3436 cm-1处出现了氨基吸收峰, 而醛基特征吸收峰消失。 在1H NMR中可以看出, δ 9.256, 7.069~7.112和6.275处出现了— NH和— NH2的化学位移, 而化合物4氢谱中醛基上氢δ 9.582~9.593的化学位移消失, 说明醛基连接了 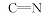

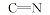
化合物7的红外光谱与化合物5的红外光谱比较, 发现1732 cm-1处的酯羰基吸收峰消失。 从该化合物的1H NMR中可以看出, δ 7.915是— NH的氢化学位移, δ 6.890~6.912为双峰, 是 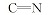
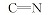
化合物8的红外光谱与化合物6的红外光谱比较, 1728 cm-1处的酯羰基吸收峰消失。 1H NMR中可以看出, 在化合物6中, 3位碳上氢化学位移为δ 4.604~4.626, 而产物8中, 3位碳上氢化学位移变为δ 3.546。 在碳谱中, 没有出现酯羰基的碳化学位移, 与此同时3位上的碳化学位移比化合物6向高场移动(71.7), δ 178.4是 
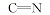
化合物4直接和氨基脲反应得到化合物5, 由于使用氨基脲的盐酸盐作为原料, 因此反应中加入乙酸钠来中和原料中的HCl, 得到游离的氨基脲分子, 然后进一步与化合物4反应。 化合物5的合成此前已有文献报道[14], 在该文献中豆甾醇臭氧化后没有分离, 直接加入氨基脲生成化合物5, 产率仅为14%。 化合物5在酸性条件下水解得到化合物7。 文中没有化合物5与7生物活性的相关报道。 在化合物6的合成中, 化合物4与硫代氨基脲的比例为1∶ 1.25, 在反应体系里滴加几滴冰醋酸调节溶液pH值为3~5, 进一步促使反应的进行。
化合物7和8的制备均采用氢氧化钾/甲醇作为反应条件, 脱去乙酰基, 得到目标产物, 该反应条件温和, 反应产率也较高。
合成化合物对6种不同肿瘤细胞株的体外抑制肿瘤细胞生长增殖活性见表1所示。 从表1数据可以看到, 甾体化合物的支链结构对化合物抑制肿瘤细胞生长增值的活性具有很大的影响。 其中具有22-缩氨硫脲支链结构的化合物8具有较好的细胞毒性, 其对MGC 7901、Hela、SMMC 7404及CNE-2细胞的IC50值与阳性对照物顺铂相当。 另外, 化合物6对Hela细胞表现了较好的选择性细胞毒性, 其IC50值达到了6.3 μ mol/L。 在这里, 母体豆甾醇没有表现出明显的细胞毒性。 对于具有22-降豆甾-22-缩氨脲支链结构的化合物7来说, 其抑制肿瘤细胞生长增殖的活性明显低于化合物8, 说明了缩氨硫脲基团是一个比缩氨脲基团更好的具有抗肿瘤活性的药效团。 同时可以看到, 当化合物的3-位羟基乙酰化后, 化合物的细胞毒性有所降低, 这可能是乙酰化后化合物的水溶度降低的缘故。
| 表1 22-含氮甾体化合物的体外抑制肿瘤细胞生长增殖活性(IC50:μ mol/L) Table 1 In vitro antiproliferative activities of 22-nitrogen-contained steroidal compounds |
从豆甾醇出发, 合成了4个22-降-豆甾-22-含氮甾体化合物, 并通过核磁共振、红外光谱和质谱对合成物进行了结构表征。 对合成产物进行体外抑制肿瘤细胞生长增殖活性研究表明, 具有22-缩氨硫脲基团的此类化合物比22-缩氨脲基团的化合物具有更好的体外抑制肿瘤细胞生长增殖活性, 其中化合物22-降-3β -羟基胆甾-5-烯-22-缩氨硫脲对MGC 7901、Hela、SMMC 7404及CNE-2细胞的抑制活性与阳性对照物顺铂相当。 上述结果为我们设计合成具有更好抗肿瘤活性的甾体化合物提供了有用的参考。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|