

GUO Hongchen, QIN Yusheng, WANG Xianhong. Acetic Acid/Oxygen Route for Efficient Synthesis of Substituted Porphyrin[J]. Chinese Journal of Applied Chemistry, 37(3): 264-270


卟啉在结构上可视为是由4个吡咯类亚甲基的 α-碳原子通过次甲基桥互联而形成的大分子杂环化合物,具有高度共轭结构。 随着卟啉与金属的配合物即金属卟啉的催化特性受到广泛关注,取代卟啉的高效合成日益显示出重要的学术和应用价值。 但是现有的丙酸法和乙酸/硝基苯法存在卟啉收率低、环境污染严重等问题,导致取代卟啉的成本居高不下,限制了其广泛应用。 本文利用乙酸/氧气法,在120 ℃下控制反应物浓度为0.24 mol/L,并在反应第一阶段(前30 min)通入氧气,得到四(4-溴苯基)卟啉(化合物d),产率高达53.8%。 该方法是高效合成取代卟啉的新方法,卟啉骨架形成和氧化两个反应同时进行,方法简单、绿色且收率高,有望大幅度降低取代卟啉的合成成本。
Recently, efficient synthesis of porphyrin has become increasingly interesting in demand of many discoveries in metalloporphyrin complexes, especially for their excellent catalytic performances. Porphyrin is a conjugated macrocyclic compound composed of four pyrrole rings connected by methenylene. The state-of-the-art porphyrin synthesis includes propionic acid method and acetic acid/nitrobenzene method, but both suffer from low yield, high cost and heavy environmental pollution. In this work, an acetic acid/oxygen route for substituted porphyrin synthesis was developed, where the reaction temperature was kept at 120 ℃, while O2 was fed only in the first 30 min under reactant concentration of 0.24 mol/L, leading to tetra(4-bromophenyl)porphyrin in the yield of 53.8%. It should be noted that the acetic acid/oxygen route allows the concurrence of macrocyclic framework formation and oxidation, which provides a viable strategy for efficient porphyrin synthesis in convenient and less environment loading way.
卟啉(porphyrin)及其衍生物在结构上可视为是由4个吡咯类亚甲基的 α-碳原子通过次甲基桥互联而形成的大分子杂环化合物,其母体为卟吩,有取代基的卟吩则称为卟啉[1,2]。 卟啉类化合物因其特殊的化学结构而具有荧光活性[3],并具有与金属配位的能力,不仅可用于分析化学[4]、医药化学[5]和光电材料化学[6,7],也是合成金属卟啉催化剂的核心原料[8,9,10]。 2008年,本课题组利用四苯基卟啉钴和双(三苯基正膦基)氯化铵(PPNCl)体系催化二氧化碳(CO2)与环氧丙烷(PO)的共聚反应[11],在25 ℃下成功实现了CO2与PO的共聚。 随着相关研究的不断深入,金属卟啉催化剂体系的不断优化[12,13,14],铝卟啉配合物已经在CO2固定高分子领域显示出巨大的工业化潜力[15,16],但是卟啉配体的高昂成本限制了其未来发展前景,因此卟啉配体的高效合成和低成本化成为必须解决的关键问题[17]。
1936年,Rothemund[18]首次用苯甲醛和吡咯做原料,以吡啶为溶剂,在密封管中加热到150 ℃,反应24 h,制备出四苯基卟啉及二氢卟啉,收率为4%~5%,当改用乙酸锌为催化剂时产率可提高到10%~11%。 不过,用这种方法合成卟啉不仅合成条件苛刻,且产率较低,只适用于少数几种芳香醛合成对应的卟啉化合物。 1964年,Alder等[19]以苯甲醛和吡咯为原料,在丙酸中加热回流0.5 h得到四苯基卟啉,产率提高到20%。 1987年,Lindsey等[20]提出用等物质的量的苯甲醛和吡咯在氮气的保护下,以二氯甲烷为溶剂,三氟化硼乙醚配合物(BF3-Et2O)或三氟乙酸(TFA)为催化剂,室温下通过缩合反应生成四苯基卟啉原,然后用二氯二氰基对苯醌(DDQ)或四氯苯醌(TCQ)将卟啉原氧化脱氢得到最终产物四苯基卟啉(TPP),产率进一步提高到30%~40%。 该方法解决了很多卟啉因Adler法条件苛刻而无法合成的问题,不过该方法也存在原料浓度过低、反应步骤较多的问题。 1994年,Lindsey等[21]提出了高浓度下的合成卟啉方案,底物浓度超过0.1 mol/mL,并可以通过一步法合成卟啉,产率也可达20%~30%,卟啉合成反应的机理(Scheme 1)为吡咯和苯甲醛先在酸的催化作用下生成卟啉原,然后卟啉原被氧化成卟啉。 1991年,郭灿城等[22]利用无水AlCl3为催化剂, N, N-二甲基甲酰胺(DMF)为溶剂,以等物质的量的吡咯和苯甲醛缩合生成TPP,产率达30%,并且反应过程无需氮气保护,产物中无副产物四苯基二氢卟啉(TPC)生成。 但该过程要在无水条件下进行,条件较苛刻,后处理也较复杂。
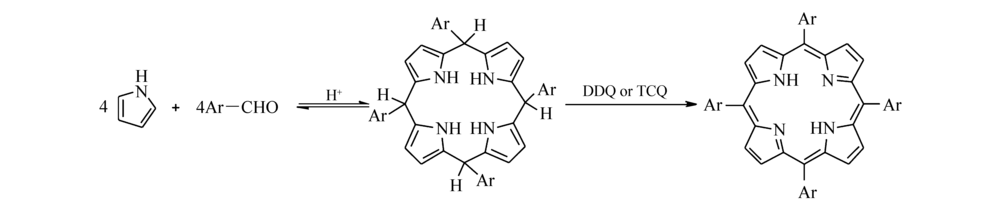 | Scheme 1 Synthetic route to porphyrin |
{kind=link}
相比于四苯基卟啉,具有取代基的四苯基卟啉因为具有更高的可修饰性,可以实现多种结构设计,因此具有更广阔的应用前景,由此高效合成取代基四苯基卟啉的研究备受关注。 Gradillas等[23]通过在合成过程中加入过渡金属盐,借助过渡金属盐的氧化作用,将合成四(4-溴苯基)卟啉的产率提高到45%。 2003年,Mamane等[24]报道了一种合成四(4-溴)苯基卟啉的方法,用醋酸和硝基苯的混合溶液代替丙酸溶液,120 ℃,反应1 h,产率达到33%,该方案反应时间短,产物收率高,但硝基苯有毒,限制了该方法的使用。
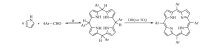
基于对成本和环境方面的考虑,希望尽量避免或减少三氟化硼乙醚、硝基苯、DDQ等价格高、污染大的试剂。 本文提出以乙酸为溶剂,利用空气或氧气直接氧化的方法制备了4种已有文献报道的卟啉化合物[25,26](Scheme 2)。 首先,将取代苯甲醛溶解在乙酸溶液中,待完全溶解后将温度升到120 ℃,然后向溶液中通入氧气或空气,滴加吡咯到乙酸溶液中后继续反应1.5 h,冷却到室温后过滤,并用甲醇和热水洗涤滤饼,干燥后称重计算产率。采用氢核磁共振波谱仪(1H NMR)确定所得产物的分子结构,并采用柱层析法确定其纯度。 本方法避免了有机强酸催化剂和硝基苯等氧化剂的使用,有可能成为一条绿色、高效合成取代基卟啉的路线。
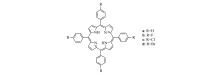
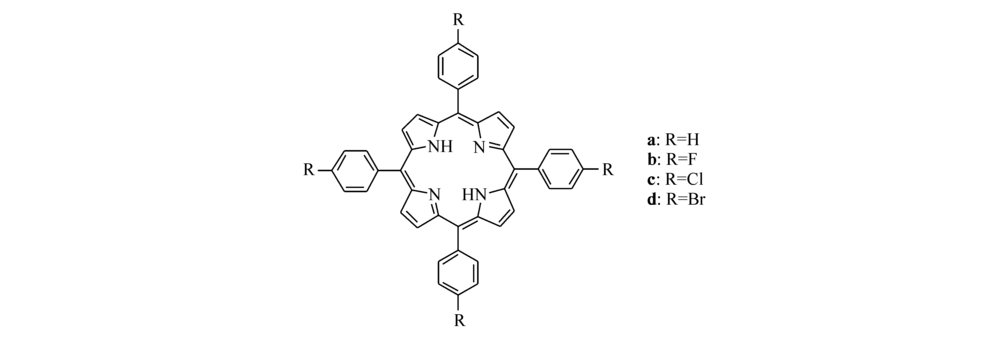 | Scheme 2 Structural formula of substituted porphyrins a-d |
{kind=link}
苯甲醛(≥99.5%)、4-氯苯甲醛(≥98%)、4-氟苯甲醛(98%)、4-溴苯甲醛(≥99%)和吡咯(≥99%)均购自阿拉丁试剂有限公司,使用前吡咯经蒸馏(Ar气保护)后待用;中性氧化铝(粒径48~75 μm,分析纯)、乙酸(分析纯)购自上海国药试剂有限公司;三氯甲烷(分析纯)、二氯甲烷(分析纯)、石油醚(分析纯)购自北京化工厂;氧气(99.5%)、空气(O2 20%,N2 80%)购自长春巨洋气体有限责任公司;CDCl3和CF3COOD购自北京伊诺凯科技有限公司。
Bruker ARX-300型核磁共振谱仪(NMR,德国布鲁克公司);Bruker AV-400型核磁共振谱仪(NMR,德国布鲁克公司);U-4100型紫外可见光谱仪(UV-Vis,日本日立公司);Bruker Autoflex Ⅲ型质谱仪(MS,德国布鲁克公司),以DCTB(2-[(2 E)-3-(4- tert-buthylphenyl)-2-methylprop-2-enylidene] malononitrile)为基质,按照反射、正离子模式测试,样品质量浓度10 mg/mL。
1.2.1 取代四苯基卟啉的合成
本文涉及的取代卟啉化合物结构式如Scheme 2所示,其中R=H,F,Cl,Br。
化合物a的乙酸/氧气法合成 在带有回流冷凝管和恒压滴液漏斗的500 mL三口瓶中加入12.72 g苯甲醛, 然后加入500 mL乙酸, 搅拌至完全溶解后升温到120 ℃, 随后将氧气或空气以40 mL/min的流速通入反应液中, 将8.30 mL吡咯在10 min内滴加到反应液中, 反应1.5 h后冷却到室温再进行过滤, 随后分别用500 mL甲醇和200 mL热水(60 ℃)洗涤滤饼, 在50 ℃下真空干燥至恒重。1H NMR(CDCl3, 300 MHz), δ : 8.89(s, 8H), 8.23~8.27(m, 8H), 7.75~7.81(m, 12H), -2.72(s, 2H); 13C NMR(CDCl3, 100 MHz), δ : 142.2, 134.6, 131.0, 127.7, 126.7, 120.1; UV-Vis(CHCl3), λ max: 419, 514, 551, 587, 646 nm; MS(MALDI-TOF) m/z calcd for C44H30N4=614.3, found 614.248(M+)。
化合物b-d的合成 按照上述类似方法合成并表征化合物b-d。
化合物b1H NMR( V(CDCl3): V(CF3COOD)=99:1), 400 MHz), δ : 8.69(s, 8H), 8.51~8.56(m, 8H), 7.72~7.78(m, 8H), -0.99(s, 2H); 13C NMR(CF3COOD, 100 MHz), δ : 134.8, 129.0, 122.4, 118.3, 112.7, 109.9; UV-Vis(CHCl3), λ max: 421, 514, 549, 590, 649 nm; MS(MALDI-TOF) m/z理论值C44H26F4N4=686.3, 测试值686.2077(M+)。
化合物c1H NMR(CDCl3, 300 MHz), δ : 8.84(s, 8H), 8.12(d, J=9 Hz, 8H), 7.76(d, J=9 Hz, 8H), -2.86(s, 2H); 13C NMR(CDCl3, 100 MHz), δ : 140.3, 135.5, 134.4, 131.1, 127.0, 118.9; UV-Vis(CHCl3), λ max: 419, 517, 550, 590, 637 nm; MS(MALDI-TOF) m/z理论值C44H26Cl4N4=750.1, 测试值750.0950(M+)。
化合物d1H NMR(CDCl3, 300 MHz), δ : 8.84(s, 8H), 8.06(d, J=9 Hz, 8H), 7.88(d, J=9 Hz, 8H), -2.86(s, 2H); 13C NMR(CDCl3, 100 MHz), δ : 140.8, 135.8, 131.9, 122.6, 119.0; UV-Vis(CHCl3), λ max: 419, 517, 553, 590, 646 nm; MS(MALDI-TOF) m/z理论值C44H26Br4N4=925.9, 测试值925.8899(M+)。
根据文献[27]报道,反应物浓度对卟啉产率和纯度有较大影响,因此系统研究了反应物浓度对合成四(4-溴苯基)卟啉(d)的影响(表1)。 合成过程中,在反应第一阶段(30 min)内通入氧气,并调整反应物吡咯和取代苯甲醛的浓度考察目标产物的收率。 由表1可知,随着反应物浓度的升高,产物收率呈现上升趋势,当反应物浓度达到0.24 mol/L时化合物d的收率为53.8%,达到最高。 但进一步提高浓度,产物收率会有所下降。因此合适的反应物浓度能够提高反应产率,浓度太低时,由于产物在乙酸中有一定的溶解性,后处理过程损失较大使得产物产率降低;浓度太高时,吡咯也可发生链增长反应生成聚吡咯伯醇,仅增加反应物浓度并不能增大产物的收率,反而增加了长链聚吡咯副产物的生成。
| 表1 反应物浓度对产率的影响 a Table 1 The influence of reactant concentration on the yield a |
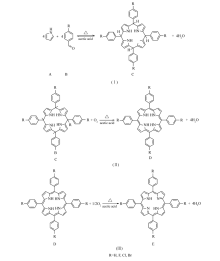
根据以上实验结果,结合Lindsey提出的卟啉合成反应机理[21],认为该反应过程主要分3步进行,如Scheme 3中(I)、(II)、(III)所示。 反应过程(I)阶段,吡咯A和取代苯甲醛B反应得到中间产物卟啉原C和水,通过将反应物A滴加到反应物B的乙酸溶液中,保证反应物B适当过量,从而有效抑制副产物的生成;进入反应(II)阶段,卟啉原C在氧气作用下被氧化成另一种中间产物即二氢取代四苯基卟吩D;到反应(III)阶段,中间产物二氢取代四苯基卟吩D在氧气作用下进一步被氧化成目标产物即取代四苯基卟啉E。
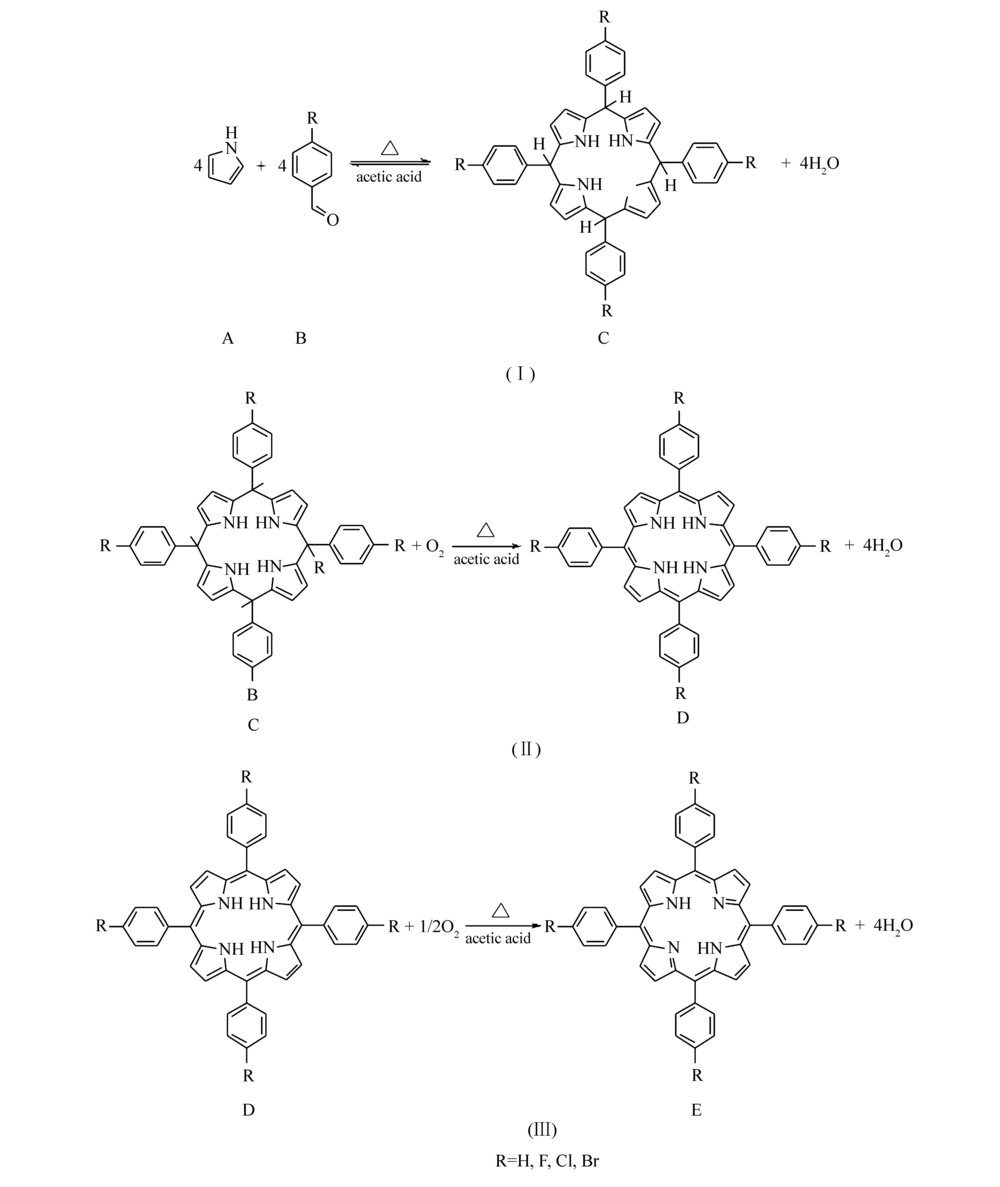 | Scheme 3 Three stages of substituted porphyrin synthesis in presence of oxygen |
{kind=link}
根据2.1节中反应物浓度对产物收率影响的研究结果,设定反应物A和B浓度为0.24 mol/L,并将反应过程均分为3个阶段,每个阶段30 min,考察在不同反应阶段通入氧气以及用空气代替氧气等实验条件变化对取代卟啉收率的影响(表2)。 由表2的Entry 1可以看出,没有氧气或空气参与反应时,产物收率只能达到25.1%;而Entries 2-6的结果表明在反应第一阶段通入氧气的收率最高,在后期进一步通入氧气,产率反而略有下降,说明反应过程中及时、适量的通入氧气能加快Scheme 3中(I)过程的正反应进程,并有效抑制副产物的生成;进一步研究结果显示,在反应第一阶段通入空气,产物收率提高并不明显。
| 表2 氧气或空气通入时间对产率的影响 a Table 2 Influence of the stage of inflowing oxygen or air on the product yield a |
在取代四苯基卟啉合成过程中,苯甲醛取代基的电子效应和位阻效应是影响合成反应产率的重要因素。 为了考察前述氧气直接氧化法对合成取代卟啉的普适性,选取前述最优反应条件(反应物A和B浓度为0.24 mol/L,反应第一阶段通入氧气),考察了取代基为H、F、Cl、Br的反应物B对反应产率的影响(表3)。表3的Entries 2-4结果显示四(4-溴苯基)卟啉产率达到53.8%,而四(4-氯苯基)卟啉和四(4-氟苯基)卟啉的产率分别下降到46.5%和38.5%,说明反应物B对位的取代基电负性越大(电负性:F>Cl>Br),产物收率越低。 苯甲醛与吡咯的缩合类似于酚醛缩合,是亲核反应,因此醛基位置电子云密度降低会使亲核试剂容易进攻,反应容易进行。 氟、氯、溴原子在苯环上是既有诱导效应又有共轭效应,电负性大的物质因为吸电子诱导效应更容易使醛基电子云密度降低,促进亲核反应的进行;但是,氟、氯、溴与苯环、醛基形成的共轭轨道是2 p、3 p、4 p轨道,显然轨道匹配程度按氟、氯、溴降低,也就是氟的孤对电子与苯环和醛基共轭程度更高,这又会一定程度上增加醛基的电子云密度,降低亲核反应的活性。 二者属于拮抗作用。 从实验结果上看,后者的作用要强于前者。
| 表3 选择不同苯甲醛对取代卟啉合成的影响 a Table 3 Effect of the selection of different benzaldehyde on the synthesis of substituted porphyrin a |
本文报道了高效合成取代卟啉的乙酸/氧气法,当反应物浓度为0.24 mol/L,在反应第一阶段(30 min)通入氧气时,取代卟啉(化合物d)收率高达53.8%。 该方法用氧气取代了如硝基苯、DDQ等传统的氧化剂,使合成过程更绿色环保,同时有助于降低反应成本。 此外,该方法合成和氧化同时进行,在短时间内即可生成目标产物,缩短了制备周期,是一种绿色、高效的取代卟啉合成方法。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|