
LIU Minghui, LIU Yingcen, L#cod#x000dc; Rongwen. Preparation of Carbon-Supported Highly Dispersed Palladium Nanoparticles and Their Performance for Suzuki Reaction[J]. Chinese Journal of Applied Chemistry, 37(1): 61-68


利用三聚氰胺甲醛预聚物中N原子与Pd2+的相互作用,将Pd2+化学锚定在预聚物中;并以二氧化硅水凝胶为造孔剂、2,4-二氨基苯磺酸为预聚物缩合促进剂,在水溶液中制备出锚定了Pd2+的胶体纳米球;再经摩尔分数5%氢气焙烧、HF腐蚀,得到Pd质量分数为1.37%、平均粒径为(2.4±0.87) nm的碳载高分散Pd纳米粒子催化剂Pd@C。 将其应用于Suzuki反应,在加入Pd与碘苯物质的量比为1:100的催化剂时,反应5 min收率为99.3%,且经8次循环后活性未降低,表现出良好的催化效果和重复使用性。
Firstly, Pd2+ ions are anchored on melamine-formaldehyde prepolymer by the coordination with N atoms on the prepolymer. Then the colloidal nanospheres grow on silica hydrogel via the simultaneous condensation reaction accelerated by 2,4-diaminobenzenesulfonic acid. Finally, the carbon-supported highly dispersed palladiumPd@C catalyst is obtained after calcination in an atmosphere of 5%(molar fraction) H2 and 95%(molar fraction) N2 gases, followed by being treated with 5% HF solution. The prepared highly dispersed Pd nanoparticles with a loading amount of 1.37%(mass fraction) have an average diameter of (2.4±0.87) nm, and the catalytic performance of Pd@C has been further evaluated by Suzuki reaction. With the addition of molar ratio 1:100 catalyst, Pd@C performs both good catalytic activity with a yield of 99.3% achieved within 5 minutes, and excellent durability after 8 cycles.





钯纳米粒子由于粒径小、比表面积大以及具有量子限域效应[1,2,3],在催化领域受到广泛关注[4,5,6]。 但正由于钯粒子粒径小,使其具有高的表面能,钯粒子极易发生团聚。 针对这一问题,人们通过将钯纳米粒子分散在载体材料上来解决[7,8,9]。 碳材料作为载体负载钯纳米粒子,相比于二氧化硅[10,11,12]、金属氧化物[13,14]等载体,具有高温稳定性好、化学惰性以及金属易回收等优点,被认为是优异的载体材料[15,16]。 负载型纳米催化剂的制备通常采用浸渍法[17]、沉积法[18]、表层反应法[19]等,但这些方法在后续还原、焙烧等阶段难以实现金属纳米粒子尺寸的有效控制、使其保持在较小的尺寸范围。 在碳载体形成的同时将钯纳米粒子原位分散,使钯粒子与载体紧密结合,是解决上述问题的有效途径。
基于此,本文在水溶液中合成三聚氰胺甲醛预聚物,利用预聚物中N原子与Pd2+相互作用,化学锚定Pd2+,预聚物进一步反应生成胶体纳米微球,并将Pd2+原位分散于胶体微球中。 将胶体微球在H2和N2混合气体(物质的量比5:95)下焙烧,得到炭载钯纳米粒子(Pd@C)纳米微球材料。 该制备过程条件温和、操作简单可控。 由于Pd2+被原位锚定在胶体微球中以及碳载体对Pd纳米粒子的稳定作用,Pd@C纳米材料中Pd粒子具有高分散性。 同时碳载体高孔隙率和化学稳定性,为Pd@C催化剂高催化活性提供可能。 在Suzuki模型催化反应中,摩尔分数1%Pd@C在5 min内获得99.3%收率。
三聚氰胺、氯亚钯酸铵、碘苯、苯硼酸、碳酸钾和乙醇均为分析纯试剂, 2,4-二氨基苯磺酸(质量分数为98%)、二氧化硅溶胶水溶液(粒径30 nm,质量分数为50%)、氢氟酸(质量分数为40%)和甲醛(质量分数为37%~41%),以上试剂均由国药集团化学试剂有限公司提供,试剂均直接使用。 实验用水均为电阻18.2兆欧的去离子水。
XD-3A型X射线衍射仪(XRD,日本岛津公司);Nova Nano-SEM 450型场发射扫描电子显微镜(SEM,美国FEI公司),加速电压5 kV;JEOL-200CX型透射电子显微镜(TEM,日本电子公司),工作电压300 kV;3H-2000PM型物理吸附仪(贝士德仪器科技(北京)有限公司); 6130MSD型液质联用仪器(HPLC-MS,美国Agilent公司);AR 2130型电子精密天平(0.0001 g,奥豪斯(上海)公司);Agilent 1100型高效液相色谱仪(HPLC,美国Agilent公司); RCT basic型恒温加热磁力搅拌器(德国IKA集团);H-1650型台式高速离心机(湖南湘仪仪器有限公司);OTL-1200型管式炉(南京大学仪器厂); Escalab 250型X射线光电子能谱仪(XPS,美国 Thermo-VG 公司),工作电压15 kV,线源为Al- KαX,使用284.6 eV的C1 s峰校准结合能及软件XPS PEAK 4.1进行曲线拟合;Optima 2000DV型电感耦合等离子发射光谱仪(ICP,美国PerkinElmer Precisely公司),其工作参数为:射频功率1300 W;辅助气流量0.20 L/min;冷却气流量15 L/min;样品提升量1.5 mL/min;积分时间10 s;观察高度15 mm。
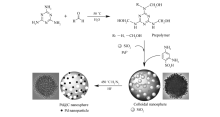
向100 mL单口烧瓶中加入0.19 g三聚氰胺(1.5 mmol)、0.5 mL甲醛溶液和40 mL去离子水,在50 ℃、500 r/min条件下搅拌至反应体系均一透明。 加入0.5 mL二氧化硅水凝胶,搅拌30 min后,准确量取0.75 mL (NH4)2PdCl4(0.5 mol/L)水溶液,加入到反应体系中,反应液呈浅黄色,继续搅拌60 min后,加入0.038 g 2,4-二氨基苯磺酸(0.2 mmol),继续搅拌反应2 h。 将反应液倒入离心管中,6000 r/min,离心10 min,20%乙醇溶液清洗后再离心,此过程重复3次,采用ICP-AES确认离心后溶液中Pd2+质量分数降低至0.01%以下,也证明Pd2+被锚定在胶体纳米微球。 将离心得到的产物在105 ℃烘干10 h,然后在摩尔分数5%氢气及95%氮气气氛下450 ℃焙烧2 h,焙烧后产物用5%氢氟酸浸泡1 h除去二氧化硅,得到催化剂Pd@C。
量取已配制的0.05 mol/L碘苯和0.05 mol/L苯硼酸乙醇溶液各10 mL,加入到50 mL圆底烧瓶中,再称量0.039 g Pd@C催化剂(Pd与碘苯的物质的量比为1:100)加入烧瓶中,搅拌下加热至体系回流后,加入0.2 g碳酸钾固体继续搅拌,并采用HPLC监测反应进程。 HPLC检测条件:流动相A为水相(含0.6%乙酸、0.3%三乙胺);流动相B为甲醇。 流动相比例为 V(A): V(B)=4:1,流速为0.8 mL/min,检测波长为(250±4) nm,进样量为5 μL。
三聚氰胺与甲醛在中性条件下、50 ℃水溶液中反应,形成三聚氰胺羟甲基化合物,常被称为预聚物;向预聚物中加入二氧化硅水凝胶及氯亚钯酸铵水溶液,预聚物中N原子与Pd2+发生相互作用[20,21,22],化学锚定Pd2+。 加入2,4-二氨基苯磺酸后,酸催化下—NHCH2OH可形成
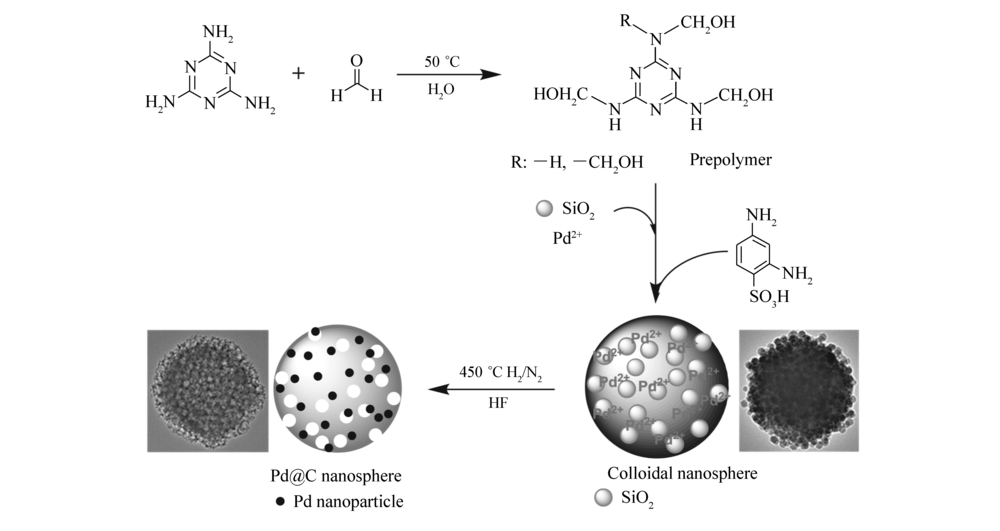 | 图1 Pd@C形成过程示意图Fig.1 Schematic illustration for the formation mechanism of Pd@C |
{kind=link}
对反应形成预聚物及Pd@C纳米材料采用HPLC-MS、SEM、TEM、XRD、XPS及BET等手段进行了表征,证明了Pd@C的形成机理和组成。
2.2.1 预聚物的结构及组成
三聚氰胺分子中含有3个氨基,每个氨基上的活泼氢原子均可与甲醛发生亲核加成反应,在甲醛量足够的情况下,可生成六羟甲基三聚氰胺。 实际反应中,三聚氰胺和甲醛反应的产物是含16个羟甲基的三聚氰胺预聚物的混合物,产物的比例受反应物的比例、反应温度、反应时间、pH值等条件的影响。 本实验中,控制三聚氰胺和甲醛的物质的量比为1:4.5,50 ℃下搅拌1 h,得到预聚物。 对该预聚物进行HPLC-MS联机分析,相对分子质量为216的三羟甲基三聚氰胺HPLC相对质量分数为56.8%,相对分子质量为246的四羟甲基三聚氰胺HPLC相对质量分数为37.1%,预聚物中三羟甲基三聚氰胺和四羟甲基三聚氰胺的含量达93.9%。 预聚物中大量的—NHCH2OH及—NH—基团,为化学锚定Pd2+及均匀分散二氧化硅水凝胶提供了保障。
2.2.2 SEM及TEM表征
酸催化条件下,预聚物分子结构中的—NHCH2OH既可生成—NHC
 | 图2 Pd-SiO2@C (A)和Pd@C(B)材料SEM照片;Pd@C材料TEM照片(C,D)(D中插图为Pd纳米粒子粒径分布图)Fig.2 SEM images of Pd-SiO2@C(A) and Pd@C(B); TEM images of Pd@C(C,D) (The inset of D shows the particle size distribution of Pd nanoparticles) |
{kind=link}
2.2.3 XPS表征
各种原子、分子不同轨道的电子结合能是一定的,具有标识性。 由于元素所处的化学环境不同,其内层电子的轨道结合能会发生变化。 因此,测定电子结合能即可进行元素的定性分析。图3A是Pd@C的XPS宽扫描谱图,显示出C1 s、N1 s、Pd3 d和O1 s的4个典型特征峰,分别对应位于285、348、399和532 eV处的峰。图3B的C1 s结合能284.6和286.2 eV可归属为C—C和C—N;图3C的N1 s的也是两个峰,它们分别对应吡啶N(398.4 eV)、吡咯N(400.2 eV)[23];图3D中Pd3 d的两个峰为335.2和340.5 eV,分别对应Pd3 d5/2和Pd3 d3/2,证明钯是Pd0形式存在[28],材料制备过程在氢气条件下还原制得,所以Pd是以零价钯存在。即所制备的Pd@C主要含有C、N、Pd和O元素,其中Pd是零价钯。
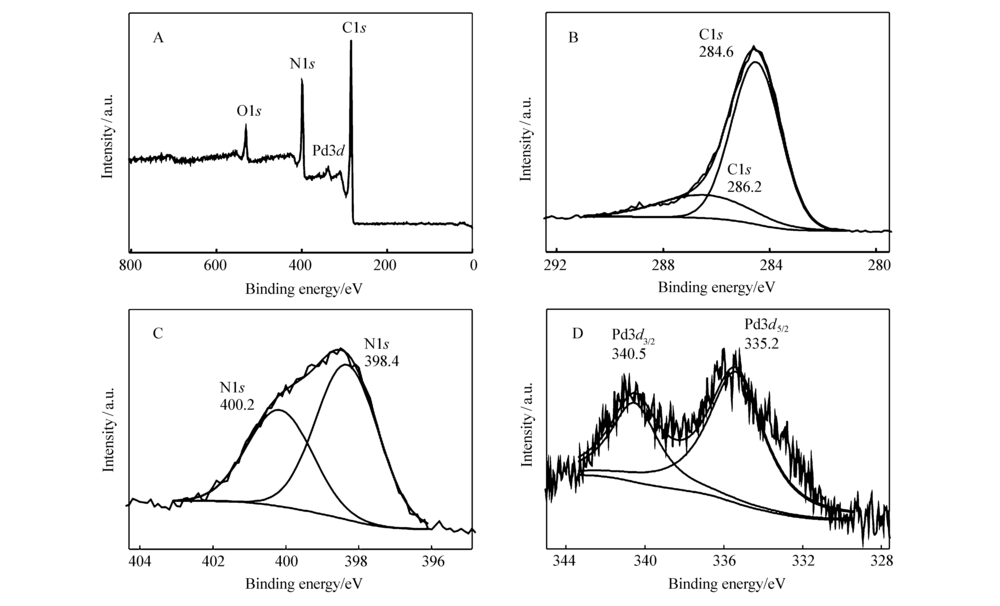 | 图3 Pd@C材料的XPS光谱宽扫描光谱(A)及C 1s (B)、N1s (C)和Pd 3d (D)的元素高分辨率光谱Fig.3 XPS survey spectra of Pd@C(A) and high resolution spectra for the elements of C1 s(B), N1 s(C), and Pd3 d(D) |
{kind=link}
2.2.4 元素分布表征
Pd@C纳米材料的元素图像分析(图4)可进一步确定Pd@C纳米材料中含有C、N和Pd 3种元素,这与XPS光谱分析结果相吻合。 同时,Pd元素在分析图像上分散均匀,也表明Pd粒子在微球上分布均匀。
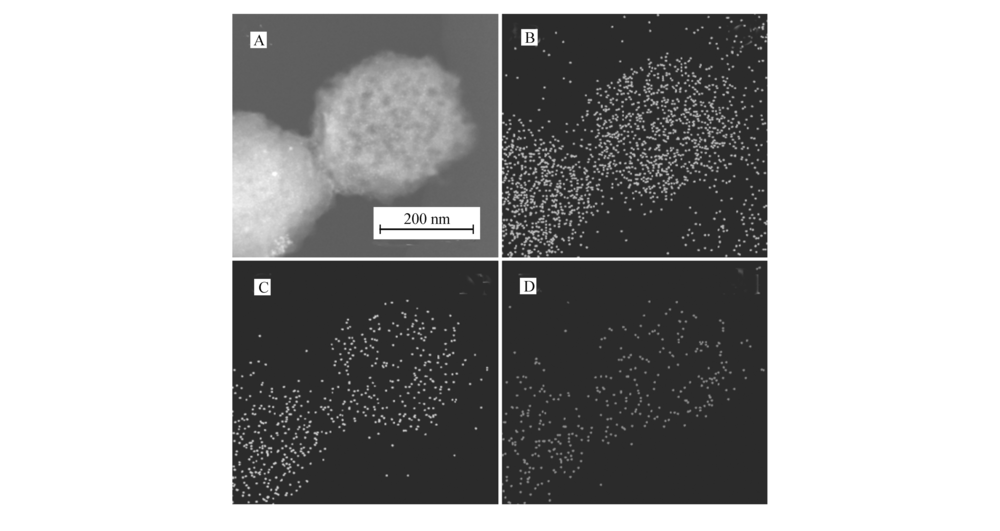 | 图4 Pd@C材料的元素分布图像: Pd@C材料的暗场扫描透射电子显微镜照片(STEM-DF)(A)、C元素(B)、N元素(C)和 Pd元素(D)Fig.4 Element mapping images of Pd@C: STEM-DF of Pd@C(A), C(B), N(C)and Pd(D) |
{kind=link}
2.2.5 XRD表征
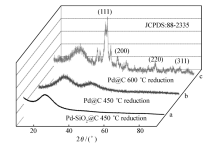
图5为纳米材料Pd@C的XRD谱图。图5谱线a和b为450 ℃还原后Pd-SiO2@C和Pd@C纳米材料的XRD谱图,未发现Pd颗粒的标准特征峰,这是由于此时Pd纳米粒子粒径比较小,衍射峰特征峰不明显,分别被二氧化硅和碳衍射峰所覆盖。 为了证明Pd@C纳米材料中Pd粒子的存在,将Pd@C纳米材料的还原温度提高到600 ℃,高温下小粒径的Pd纳米粒子将进一步聚集,形成Pd大颗粒。图5谱线c为600 ℃还原后Pd@C纳米材料的XRD,此时可以清晰的看到出现在40°、46°、68°和82°的Pd颗粒的特征衍射峰,分别对应Pd的(111)、(200)、(220)和(311)晶面,参照标准卡片(JCPDS:88-2335),表明纳米材料中Pd纳米粒子粒径较小,这是由于Pd2+被化学锚定在胶体微球中及碳对Pd纳米粒子的稳定作用。
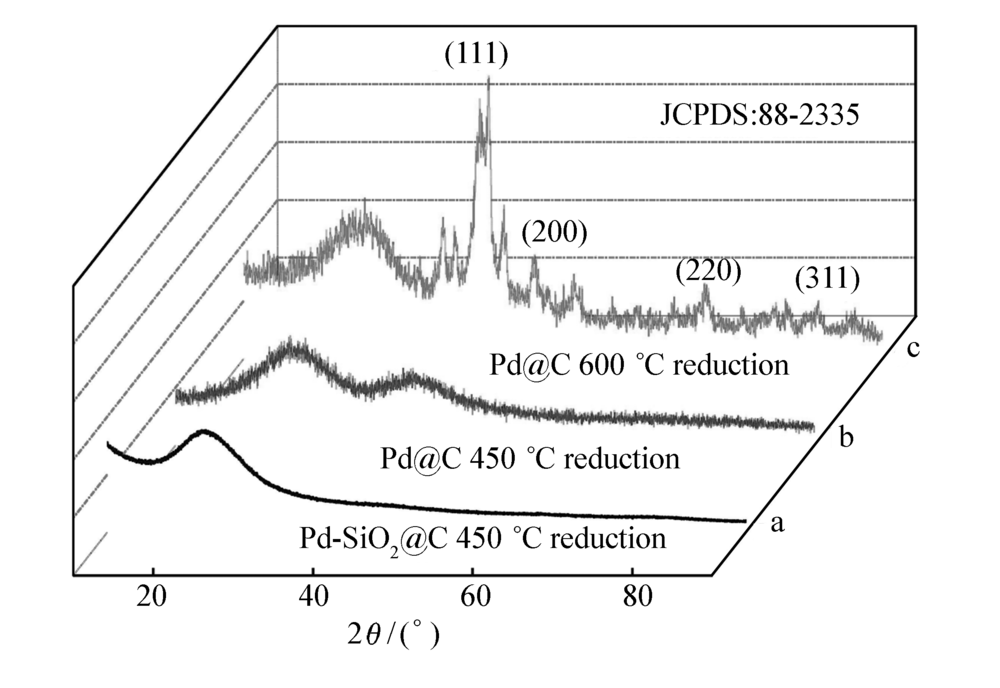 | 图5 Pd@C材料XRD谱图Fig.5 XRD patterns of Pd@C |
{kind=link}
2.2.6 BET表征
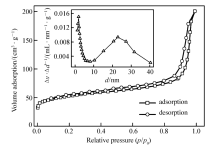
Pd@C纳米材料的氮气吸附-脱附等温曲线及孔径分布如图6所示。 根据BET计算结果,Pd@C材料的比表面积为169.3 m2/g,总孔体积为3.2 m3/g,平均孔径为24.74 nm,这与TEM图像中观察到刻蚀二氧化硅后形成的孔径基本相符,且为多孔结构。 这种孔结构的存在有利于Pd纳米粒子与反应物接触,有利于Pd纳米粒子的催化性能。
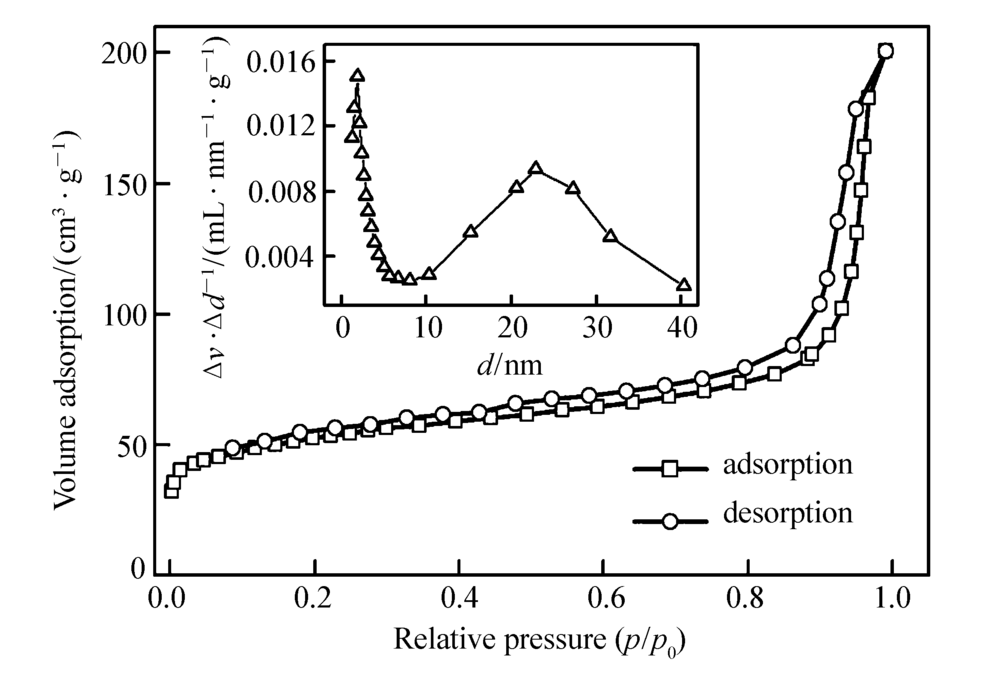 | 图6 Pd@C材料的氮气吸附-脱附曲线及孔径分布图Fig.6 Nitrogen adsorption-desorption isotherms of Pd@C(The inset shows the pore size distribution of Pd@C) |
{kind=link}
以Suzuki偶联反应为模型反应,测试Pd@C纳米材料的催化性能[29],研究了反应时间、催化剂用量及催化剂循环使用对产物收率的影响,Scheme 1为Suzuki偶联反应的条件及反应方程式。
 | Scheme 1 Suzuki cross-coupling reaction of iodobenzene with phenyl boronic acid |
{kind=link}
Pd@C催化剂用量选用与碘苯物质的量比为1:100,用已配制的碘苯与苯硼酸乙醇溶液进行Suzuki反应。 分别在1、2、3、4、5、6和7 min时取样,进行液相色谱分析。 结果表明(如图7A),随着时间的增加,收率逐渐增大,反应1 min时,收率为22.7%;反应5 min时,收率可达到99.3%,反应时间进行到6和7 min时,收率增大不明显。 表明Pd@C纳米材料在5 min即可催化Suzuki反应完成。
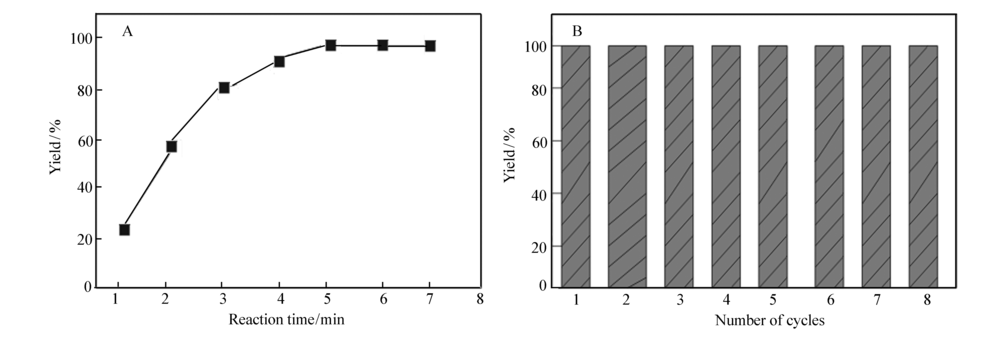 | 图7 反应时间对收率的影响(A)和Pd@C催化Suzuki反应循环性能图(B)Fig.7 Effect of reaction time on yield(A) and cyclability tests of the Pd@C for catalytic Suzuki reaction(B) B.The yield was measured after 5 min reaction |
{kind=link}
在其它条件不变的情况下,通过控制催化剂的用量来探讨其对反应速率的影响,Pd@C催化剂用量分别取用与碘苯物质的量比0.3:100、0.5:100、1:100、1.5:100和2:100进行Suzuki反应,均在5 min时取样。 液相检测分析结果表明:Pd@C催化剂用量摩尔分数在0.3%、0.5%、1%时,随着催化剂的用量的增加,收率增大;而催化剂与碘苯物质的量比在1:100、1.5:100和2:100时,收率基本相同,即与碘苯物质的量比1:100是催化剂的合适用量。
以产物收率为考察指标,Pd@C催化剂用量与碘苯物质的量比为1:100 时,反应5 min,收率为99.3%。 催化剂循环使用8次,反应5 min时,收率未见明显下降,保持在98%以上(图7B),说明所制备的Pd@C催化材料对碘苯与苯硼酸的Suzuki偶联反应具有较好的循环使用稳定性。
本文合成了一种碳载高分散Pd纳米粒子催化剂Pd@C。利用三聚氰胺甲醛预聚物中N原子与Pd2+配合作用,将Pd2+原位锚定在预聚物中,加入二氧化硅水溶胶造孔,通过2,4-二氨基苯磺酸促进预聚物聚合,形成Pd2+均匀分布的胶体纳米球;再经过焙烧还原及HF刻蚀除去SiO2,得到Pd@C纳米材料。 基于N原子对Pd2+的化学锚定及碳载体对Pd纳米粒子的稳定作用,Pd纳米粒子均匀分布在碳球中,平均粒径为(2.4±0.87) nm。 在Suzuki模型催化反应中,与反应物物质的量比为1:100的 Pd@C在5 min内获得99.3%的收率,证明Pd@C纳米材料在催化方面具有较好的应用前景。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|