
ZENG Huijuan, LIN Meijuan, LIU Chao, et al. White Phosphorescent Star-Shape Polymers Derived from Poly(fluorene-carbazole) with Green-Yellow Iridium Complex as the Cores[J]. Chinese Journal of Applied Chemistry, 37(1): 16-23


设计合成了以苯基苯并咪唑和吡啶三唑为配体的高效的黄绿光铱配合物(M1),并通过Suzuki缩聚反应制备了以磷光铱配合物客体为中心核、蓝光荧光聚(芴-咔唑)主体为臂的星型磷光聚合物(P2.5、P5.0和P10),着重对M1和聚合物的发光性能、电化学性能及热稳定性能进行研究。 结果表明,M1具有较高的荧光量子效率(32.06%),其荧光寿命为1.09 μs,聚合物荧光寿命为2.223.93 μs,均表现为磷光;通过调节主客体的比例,利用主客体的部分能量转移机制,来实现聚合物的不同光色,发光颜色可从蓝光向黄光变化;当M1摩尔分数为2.5%时,获得的白光聚合物(P2.5)具有较好的发光性能和热稳定性能,色坐标为(0.30,0.32),位于白光区域,其最高占有轨道(HOMO)能级和最低未占有轨道(LUMO)能级分别为5.49和2.43 eV,荧光量子产率为14.3%,荧光寿命为2.22 μs。
An efficient green-yellow light iridium complex based on 2-(4-brominephenyl)-1-octyl-benzimidazole as cyclometalated ligand and 3-bromine-5-(pyridin-2-yl)-1 H-1,2,4-triazole as ancillary ligand was synthesized. A series of novel star-shape phosphorescent polymers (P2.5, P5.0 and P10) was synthesized by employing the iridium complex as the core guest and the poly(fluorine-carbazole) as the arm host through Suzuki cross-coupling. The properties of the iridium complex and polymers were studied. The results showed that the iridium complex emitted green-yellow light with peaks at 490,526 and 565 nm and its fluorescent quantum efficiency was 32.06%. Fluorescent lifetimes for the iridium complex and the polymers are in the microsecond regime (1.093.93 s). Such long-lived excited states clearly suggest that the emitting state has triplet phosphorescent emission. The yellow light intensity was enhanced with the increasing of iridium complex content, indicating that there exists partial energy transfer from host to guest, and the emission color of polymers shifts from blue to yellow by adjusting the proportion of the iridium complex. When the mole fraction of iridium complex reached to 2.5%, the white-light polymer (P2.5) was obtained, the CIE1931 chromaticity coordinate was (0.30, 0.32), the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels were 5.49 and 2.43 eV respectively, fluorescent quantum efficiency was 14.3%, and fluorescent lifetime was 2.22 s. The polymer having good thermal stability can satisfy the requirements of luminescent materials.

在发光材料中,三线态能级较高且具有优良载流子传输性能的含芴或咔唑为基本结构单元的小分子或其聚合物等常作为主体材料,发光金属配合物常作为客体材料,主客体通过掺杂或键合在一起,利用它们之间的能量传递来调节光色以及提高发光效率[1,2,3,4,5,6]。 将发光金属配合物键入到线型聚合物主链或支链上或作为星型聚合物发光中心可以进一步解决掺杂体系的相分离问题。 与线型的聚合物相比,星型聚合物具有独特的三维支化构象,可有效地避免分子链间强烈的相互作用,能够克服主链型聚合物可能存在的 π-π堆积导致发光效率下降、以及侧链型客体发光单元存在碰撞猝灭而影响到客体发光效率下降等不足,有利于发色团高效发挥其性能,同时高度支化的结构使聚合物具有优良的溶解性和成膜性能,且利用客体外围的主体臂单元将中心核铱配合物保护起来,有效避免了浓度猝灭[7,8,9,10,11,12]。 通常采用红、绿、蓝三基色组合或者蓝、黄/橙互补色组合的方法来实现白光发射[13,14,15],尤其是二元互补色实现白光更加简单。 部分能量转移机理作为一种便捷高效的方式,已经成为研究制备白光有机发光材料的主流策略。 与传统的荧光材料相比,磷光材料能够同时利用单重态和三重态激子,具有更高的内量子效率[16]。 本文设计合成了以2-(4-溴苯基)-1-辛基-苯并咪唑为环金属配体、以3-溴-5-(吡啶-2-基)-1 H-1,2,4-三唑为辅助配体的高效的黄绿光磷光铱配合物(M1),并作为中心核、以蓝光荧光聚芴-咔唑为臂合成了星型聚合物(P2.5、P5.0和P10),通过能量不完全转移机理,以期实现白光星型磷光聚合物。
水合三氯化铱(铱质量分数>58%)购自上海久岭化工有限公司;2-(4-溴苯基)-1-氢-苯并咪唑(≥98%)购自百灵威科技有限公司;碘辛烷(>97%)购自东京化成工业株式会社;2,7-二溴-9,9-二辛基芴(≥98%)、2,7-双(4,4,5,5-四甲基-1,3,2-二氧杂硼烷-二基)-9,9-二辛基芴(≥98%)、3,6-二溴- N-辛基咔唑(≥98%)购自深圳睿讯光电材料科技有限公司;四三苯基膦钯,分析纯,购自上海阿拉丁科技股份有限公司。
ARX 400型核磁共振波谱仪(NMR,德国Bruker公司);UV-Vis-2600型紫外可见分光光度计(UV-Vis,日本岛津公司);FLS920型瞬态/稳态荧光光谱仪(英国爱丁堡公司);TGA/SDTA 851型傅里叶变换红外-热重分析联用仪热重分析仪(FTIR-TGA,瑞士Mettler-Toledo公司);1515型泵凝胶色谱仪(GPC,美国Waters公司),用标准的聚苯乙烯作为参考样,四氢呋喃作为淋洗液;CHI600B型电化学工作站(上海辰华仪器公司),选择0.1 mol/L的六氟磷酸四丁基季胺盐的乙腈溶液为电解质,工作电极为玻碳电极,铂电极为辅助电极,Ag/AgCl选为参比电极。
铱配合物(M1)单体的合成路线如Scheme 1所示。
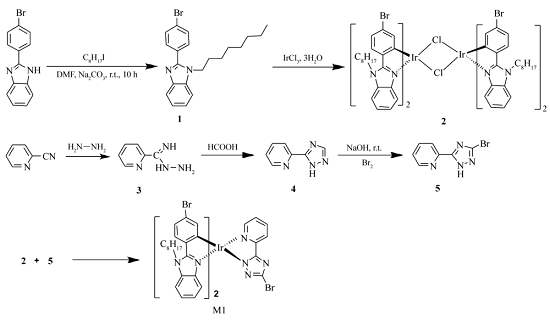 | Scheme 1 Synthesis of iriduim complex M1 |
{kind=link}
2-(4-溴苯基)-1-辛基-苯并咪唑(1) 在圆底烧瓶中加入0.82 g(3.0 mmol)2-(4-溴苯基)-1-氢-苯并咪唑和25 mL N, N-二甲基甲酰胺,搅拌溶解,再加入1.25 g无水碳酸钠和0.96 g(4.0 mmol)碘辛烷,室温反应10 h。 反应结束后,将反应混合物倾入乙酸乙酯和蒸馏水的混合溶剂中,萃取,合并有机相,并分别用饱和食盐水和2 mol/L NaOH洗涤3次,用无水MgSO4干燥,旋蒸去除溶剂,用 V(石油醚): V(乙酸乙酯)=4:1作为淋洗剂,进行柱层析分离提纯,得到0.82 g 2-(4-溴苯基)-1-辛基-苯并咪唑淡黄色油状物,其产率为71.0%。1H NMR(400 MHz,CDCl3), δ:7.847.78(m,1H),7.697.64(m,2H),7.617.56(m,2H),7.41(dt, J=4.5,2.7 Hz,1H),7.30(ddd, J=15.8,7.6,4.6 Hz,2H),4.244.16(m,2H),1.841.74(m,2H),1.291.18(m,10H),0.86(dd, J=9.4,4.5 Hz,3H)。
2-(4-溴苯基)-1-辛基-苯并咪唑氯桥联二聚体(2) 参考文献[17],用IrCl3·3H2O与2-(4-溴苯基)-1-辛基-苯并咪唑(摩尔比为1:21:2.5)在乙二醇单乙醚和蒸馏水(体积比3:1)的混合溶剂中120 ℃下回流反应24 h制得。 将所得黄色固体粉末氯桥联二聚体直接密封保存,不需要通过进一步提纯。
2-吡啶脒腙(3)[2] 在圆底烧瓶中加入5.2 g(50 mmol)2-氰基吡啶、3.4 g(55 mmol)80%水合肼和5 mL无水乙醇,室温搅拌6 h,有大量沉淀生成,减压抽滤,用石油醚多次洗涤滤饼,得到黄白色块状固体,真空干燥24 h,产率75%。1H NMR(400 MHz,CDCl3), δ:8.51(ddd, J=4.9,1.6,1.0 Hz,1H),8.00(dt, J=8.1,1.0 Hz,1H),7.747.63(m,1H),7.287.25(m,1H),5.27(s,2H),4.57(s,2H)。
3-(吡啶-2-基)-1 H-1,2,4-三唑(4) 冰浴搅拌下,将3.7 g(27 mmol)2-吡啶脒腙溶于10 mL甲酸,再缓慢滴加30 mL甲酸,室温反应2 h后,再升温到115 ℃回流反应2 h。 反应结束,冷却至常温,用50%NaOH溶液调节pH值为67,分别用乙酸乙酯溶剂和饱和食盐水各萃取3次,无水MgSO4干燥24 h,三氯甲烷重结晶得到白色固体,产率为60%。1H NMR(400 MHz,CDCl3), δ:13.02(s,1H),8.838.71(m,1H),8.26(d, J=7.9 Hz,1H),8.14(s,1H),7.91(td, J=7.7,1.0 Hz,1H),7.517.38(m,1H);13C NMR(101 MHz,CDCl3), δ:155.01,150.47,149.47,146.67,137.85,125.01,122.03。
3-溴-5-(吡啶-2-基)-1 H-1,2,4-三唑(5) 在圆底烧瓶中加入4.38 g(30 mmol)3-(吡啶-2-基)-1 H-1,2,4-三唑和75 mL蒸馏水,搅拌,溶液呈白色浑浊后缓慢滴入20 mL 10 mol/L NaOH溶液,白色固体逐渐溶解,用NaOH溶液维持pH值在12左右,用注射器滴加3 mL液溴,常温下反应4 h后用5 mol/L盐酸调节溶液pH值到34,有白色絮状沉淀析出,抽滤,真空干燥24 h后得到白色固体产物4.7 g,产率为70%。1H NMR(400 MHz,DMSO), δ:13.05(s,1H),8.71(d, J=4.1 Hz,1H),8.02(d, J=8.0 Hz,2H),7.56(dd, J=6.8,5.0 Hz,1H)。
铱配合物(M1) N2气保护下,在圆底烧瓶中加入0.20 g(0.1 mmol)2-(4-溴苯基)-1-辛基-苯并咪唑氯桥联二聚体(2),0.056 g(0.25 mmol)3-溴-5-(吡啶-2-基)-1 H-1, 2,4-三唑(5),70 mg无水碳酸钠和15 mL乙二醇单乙醚,120 ℃回流反应24 h。 冷却,水洗,过滤,所得沉淀依次用乙醇、乙醚、正己烷洗涤,得到的粗产物用二氯甲烷作为洗脱剂进行柱层析纯化,获得黄绿色固体粉末铱配合物(M1),产率为39.6%。1H NMR(400 MHz,CDCl3), δ:8.158.11(m,1H),7.907.86(m,1H),7.83(td, J=7.7,1.6 Hz,1H),7.607.50(m,2H),7.37(t, J=7.7 Hz,2H),7.30(dd, J=7.2,1.0 Hz,1H),7.287.18(m,3H),7.11(dddd, J=10.4,9.3,7.0,1.7 Hz,3H),6.866.81(m,1H),6.51(d, J=2.0 Hz,1H),6.35(dd, J=5.1,3.1 Hz,2H),4.714.50(m,4H),2.071.94(m,4H),1.461.25(m,20H),0.89(dt, J=7.4,3.8 Hz,6H)。
通过Suzuki缩聚反应合成聚合物(P2.5、P5.0和P10),合成路线如Scheme 2所示。
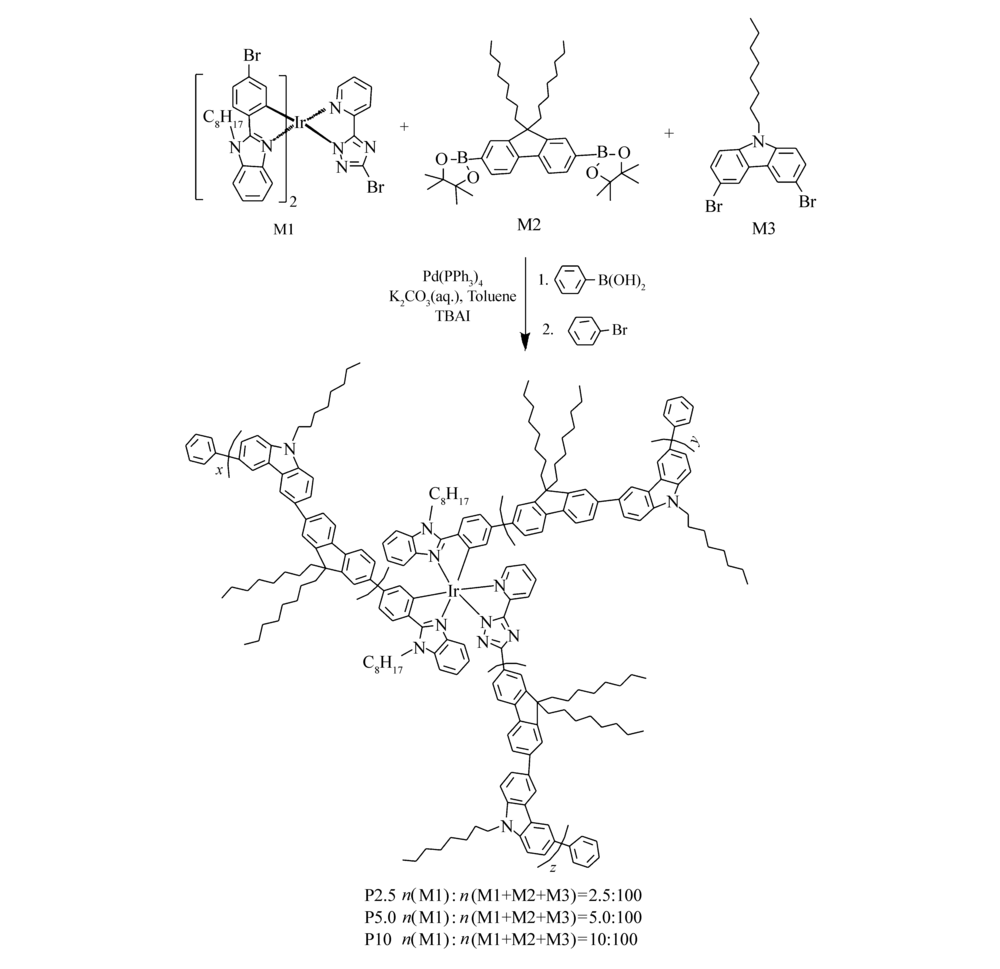 | Scheme 2 Synthesis of P2.5, P5.0 and P10 polymers |
{kind=link}
P2.5的具体合成步骤如下:向预先充分充氮除氧的2,7-双(4,4,5,5-四甲基-1,3,2-二氧杂硼烷-二基)-9,9-二辛基芴(M2)(128 mg,0.2 mmol)、四三苯基膦钯Pd(PPh3)4(12 mg,0.01 mmol)、四叔丁基碘化铵(46 mg,0.12 mmol)、M1(1.8 mg,0.01 mmol)和3,6-二溴- N-辛基咔唑(M3)(83.1 mg,0.19 mmol)的密闭混合体系中注入K2CO3溶液(2 mol/L, 2.5 mL)和6 mL甲苯,N2气气氛下110 ℃回流反应48 h,用0.11 g苯硼酸封端反应12 h后,再加入0.1 mL溴苯封端反应12 h后,冷却到室温,逐滴加入到 V(甲醇): V(水)=9:1的混合溶液中,有絮状沉淀析出,过滤,依次用甲醇、丙酮索提除去未反应的小分子和齐聚物,再用三氯甲烷索提收集聚合物,所得溶液浓缩至适当浓度后用甲醇沉析,过滤,得到淡黄色固体聚合物P2.5,M1的摩尔分数为2.5%,真空干燥24 h,产率68%。1H NMR(400 MHz,CDCl3), δ:8.50,7.857.72,7.587.52,7.006.98,4.39,3.49,2.262.12,2.021.96,1.571.11,0.900.76; Mn:2.51×105,相对分子质量分布(PDI) 2.20。
采用类似的合成手段,合成出含有M1摩尔分数分别为5.0%和10%的P5.0和P10。
P5.0:产率71%;1H NMR(400 MHz,CDCl3), δ:8.50,7.857.70,7.597.52,7.016.96,4.39,3.49,2.262.12,2.021.96,1.571.11,0.900.74; Mn:2.98×105,PDI 2.47。
P10:产率66%;1H NMR(400 MHz,CDCl3), δ:8.50,7.857.72,7.587.52,7.026.97,4.393.69,2.272.10,2.021.98,1.571.10,0.900.77; Mn:2.97×105,PDI 2.54。
所制备的聚合物在甲苯、三氯甲烷、四氢呋喃等常见的有机溶剂中具有良好的溶解性,通过溶液旋涂获得聚合物薄膜表面平整光滑。
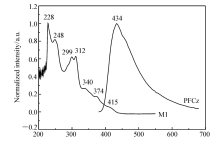
图1为M1在CHCl3溶液中的紫外吸收光谱以及聚(芴-咔唑)PFCz薄膜的荧光发射图。 M1的紫外吸收谱图中位于228、248、299和312 nm的吸收峰可归属于主配体的 π→ π*跃迁吸收。340、374 nm处吸收峰可归属于M1自旋允许的金属到配体的单重态电荷转移跃迁(1MLCT)以及配体到配体的单重态电荷转移跃迁(1LLCT)。 415 nm处吸收峰为金属到配体三重态电荷转移跃迁以及3 π→ π*三重激发态的跃迁吸收[18]。 聚(芴-咔唑)主体的荧光发射峰位于434 nm处,其荧光发射光谱和磷光M1的紫外吸收光谱在380440 nm范围内有一定的重叠,表明聚合物主体PFCz与磷光M1客体之间可发生有效的Foster能量转移。
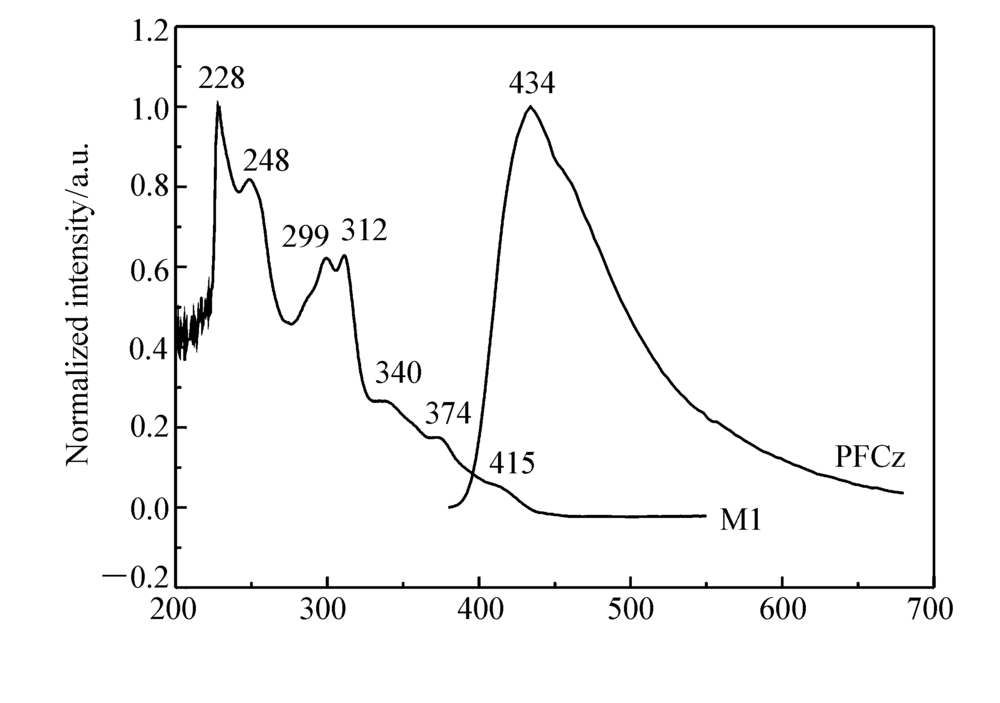 | 图1 铱配合物M1在CHCl3中紫外吸收光谱与聚(芴-咔唑)薄膜荧光发射光谱Fig.1 UV-Vis absorption spectra of iridium complex M1 in CHCl3 and PL spectrum of the PFCz film |
{kind=link}
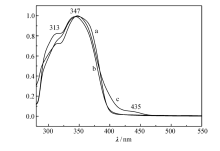
图2为聚合物在三氯甲烷溶液中的紫外吸收光谱图。图2中所有聚合物均具有相似的吸收图谱,最大吸收峰位于347 nm附近,主要体现主体PFCz的特征吸收。 随着M1摩尔分数的提高,位于313和435 nm附近的吸收峰逐渐明显,呈现客体M1的特征吸收。
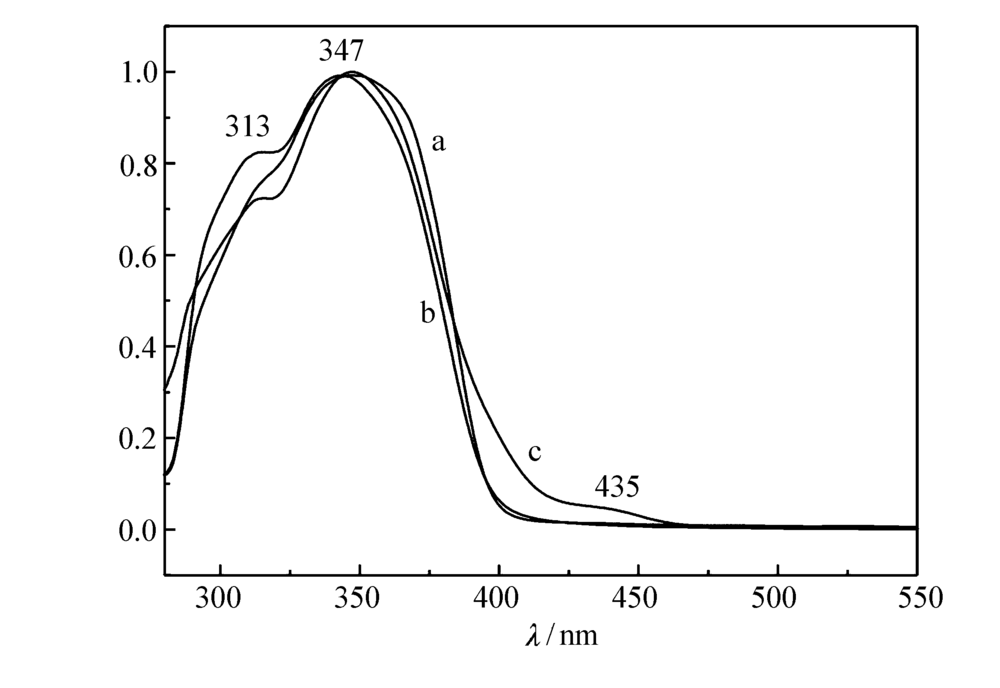 | 图2 P2.5(a)、P5.0(b)和P10(c)在CHCl3溶液中的紫外吸收光谱Fig.2 UV-Vis absorption spectra of P2.5(a), P5.0(b) and P10(c) in CHCl3 |
{kind=link}
图3为M1和星型聚合物在三氯甲烷溶液中和粉末/薄膜状态下的光致发光图。 由图3可见,M1在溶液和粉末状态下的荧光发射基本相似,发射峰位于490、526和565 nm(肩峰),为黄绿光发光,与溶液状态相比,固态下的荧光发射峰略有红移。 聚合物在溶液状态下的发射峰位于420 nm附近,仅体现为聚合物主体芴和咔唑的发射峰,而未出现客体M1发射峰,说明溶液状态下分子内聚合物链中主体的能量未充分转移给客体M1。 而在薄膜状态下聚合物的荧光发射与溶液状态有较大的不同,出现了客体M1的荧光发射峰,且随着M1摩尔分数的提高,主体的发射峰逐渐减弱,客体发射峰逐渐增强,显示了主体臂支化单元向客体中心核单元的能量传递。 这是由于能量传递与发光单元间的距离有关,固体状态下,分子链间、分子链中的主体和客体之间距离较小,因此分子内和分子间同时发生从聚合主体到磷光M1客体间有效的Foster能量转移,随着M1摩尔分数的增加,能量从聚合主体转移给客体M1更加彻底。 当配合物摩尔分数为2.5%和5.0%时,聚合物P2.5和P5.0呈现聚合物主体和M1客体的双极发射,当M1摩尔分数为10%时,主体蓝光发射峰完全消失,仅体现中心核M1位于560和603 nm的发射峰,与M1的最大发射峰528 nm相比发生了红移,这可能是因为聚合物的共轭长度增加所导致。
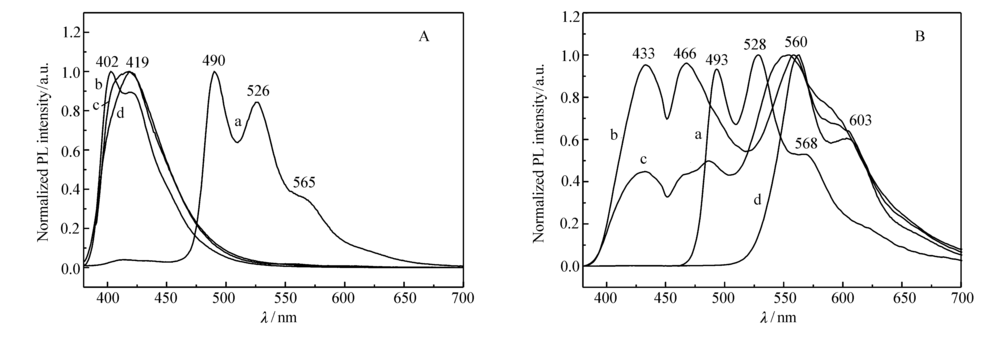 | 图3 M1(a)、P2.5(b)、P5.0(c)和P10(d)在CHCl3溶液(A)和粉末/薄膜状态(B)的荧光光谱Fig.3 PL spectra of M1(a), P2.5(b), P5.0(c) and P10(d) in CHCl3(A) and in powder/film(B) |
{kind=link}
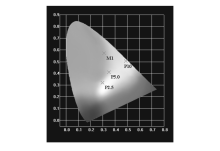
通过1931CIE色坐标拟合得到M1色坐标值为(0.31,0.57),位于黄绿光区域;而对于聚合物体系,由于臂支化单元向中心核单元的能量传递,使不同M1摩尔分数的聚合物体系呈现不同的发射光谱,从而呈现了不同的发光颜色,体现了聚合物不同的色坐标。 从发射谱图看出,当M1摩尔含量为2.5%时,P2.5呈现主体和客体的双极发射,显示客体间能量传递不完全,发光波长从蓝光到红光的覆盖,通过色坐标拟合,聚合物P2.5的色坐标值为(0.30,0.32),为白光发射,与饱和白光的色坐标(0.33,0.33)较为接近。 随着M1的比例的增加,发光颜色逐渐变成黄光,其中P5.0色坐标为(0.35,0.41),位于黄光与白光的边界,P10色坐标为(0.49,0.51),位于黄光区域。 M1和P2.5、P5.0和P10的色坐标如图4所示。
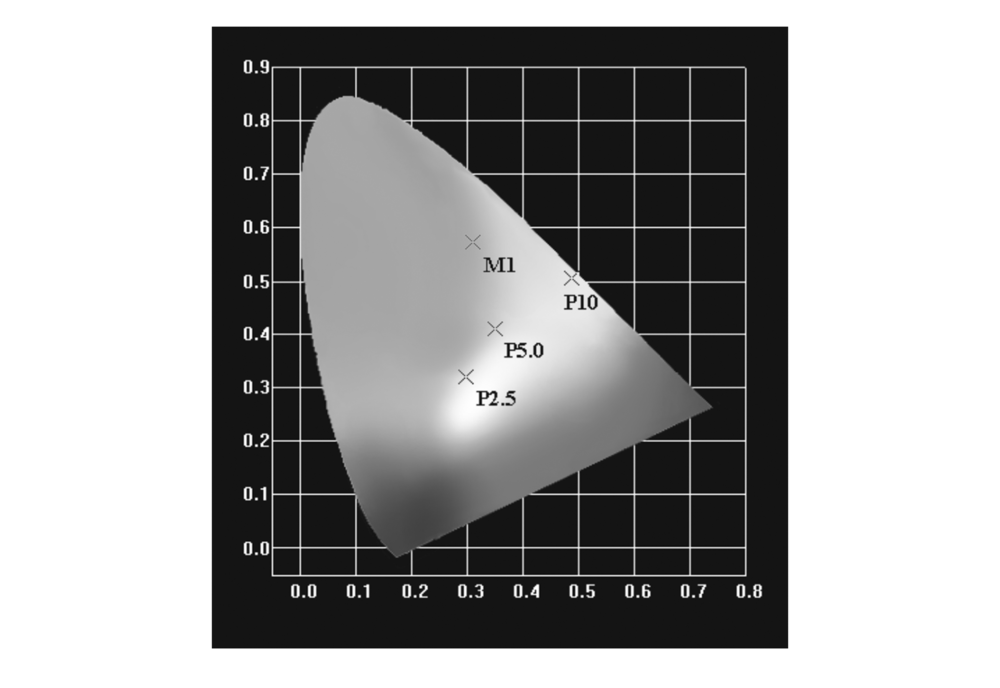 | 图4 M1、P2.5、P5.0和P10的1931CIE色坐标图Fig.4 CIE1931 chromaticity coordinates for M1, P2.5, P5.0 and P10 |
{kind=link}
以氙灯作为激发光源,选择聚合物(配合物)的最佳激发波长318 nm(365 nm)作为检测波长,在积分球中测定聚合物和配合物粉末量子产率。 M1荧光量子效率高达32.06%,与乙酰丙酮或邻菲罗啉为辅助配体相比[18],3-溴-5-(吡啶-2-基)-1 H-1,2,4-三唑为辅助配体的M1具有更优异的发光性能。 P2.5、P5.0和P10的荧光量子效率分别为14.3%、7.93%和5.55%,即随着配合物在聚合物中摩尔比例的增加,聚合物的荧光量子效率呈下降趋势。 这与文献[19]描述一致,可能是由铱的重原子效应所引起的。
在室温条件下,对M1粉末和聚合物在薄膜状态下的荧光强度衰减曲线进行表征,表明配合物和聚合物材料荧光强度的衰减均符合多指数函数 F( t)= A+ B1exp(- t/τ1)+ B2exp(- t/τ2),式中, τ表示荧光寿命(μs),B表示相对强度(%),拟合度用 χ2表示,通过计算公式 τ0=∑ τi× Bi%可以得出配合物荧光衰减寿命为1.09 μs,聚合物P2.5、P5.0、P10荧光衰减寿命分别为2.22、3.41和3.93 μs,均为微秒级别,显示三线态特征,符合磷光材料寿命衰减的特点。 M1以及聚合物光物理性能数据归纳于表1。 从表1的 kr/ knr可知,与M1相比,聚合物的非辐射程度提高,且随着M1摩尔分数的增加,铱的重原子效应导致发光效率地下降。
| 表1 M1和聚合物的光物理性能数据 Table 1 Photophysical properties of M1, P2.5, P5.0 and P10 |
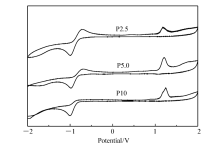
聚合物的起始氧化电势通过循环伏安测试进行表征,以玻碳电极为工作电极、Ag/AgCl为参比电极,循环伏安曲线(CV)如图5所示。 聚合物起始氧化电势为1.071.09 V,结合其紫外吸收边,计算出带隙 Eg、最高占有轨道(HOMO)和最低未占有轨道(LUMO)能级,结果如表2所示。 所有星型聚合物HOMO能级为-5.47-5.49 eV,LUMO能级为-2.36-2.43 eV,说明M1的摩尔分数对聚合物的HOMO能级和LUMO能级影响不大。
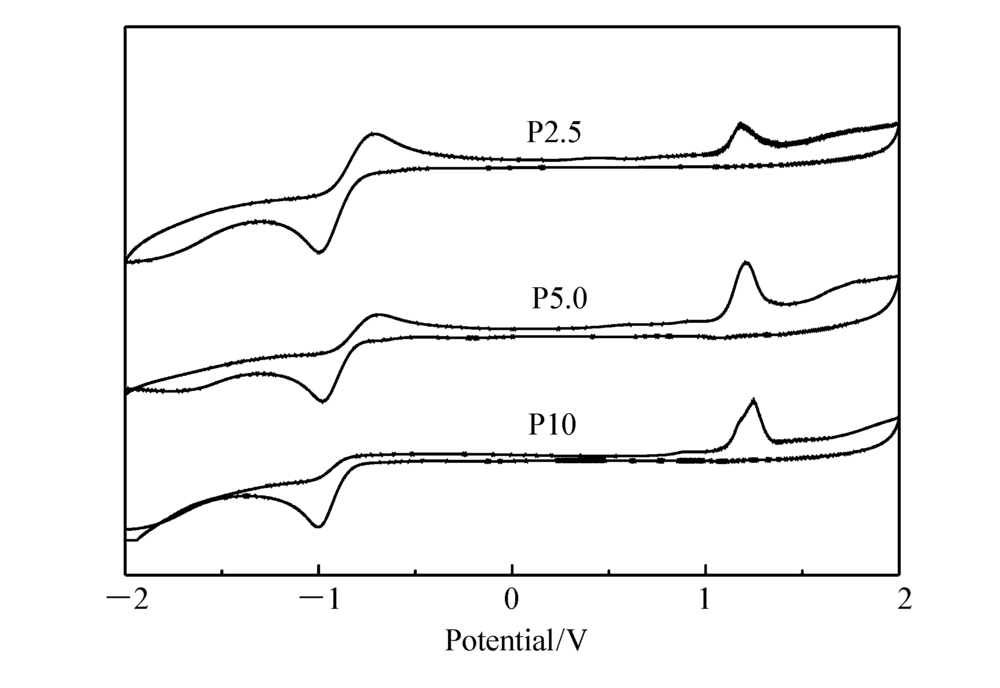 | 图5 P2.5、P5.0和P10在乙腈溶液中的循环伏安曲线图Fig.5 The CV curves of P2.5, P5.0 and P10 in acetonitrile solution |
{kind=link}
| 表2 共聚物的电化学性能数据 Table 2 Electrochemical properties of P2.5, P5.0 and P10 |
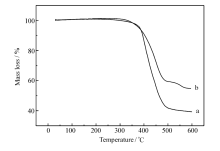
良好的热稳定性是作为发光材料的一个重要性能。 通过TGA对P2.5和P10进行了热分析表征,如图6所示。 P2.5和P10在失重5%时所对应的热分解温度分别为383和381 ℃, 失重10%时所对应的温度分别为395和403 ℃,说明聚合物具有优良的热稳定性,且随着配合物摩尔分数的提高,聚合物的热稳定性进一步提高,能够满足发光材料的要求。
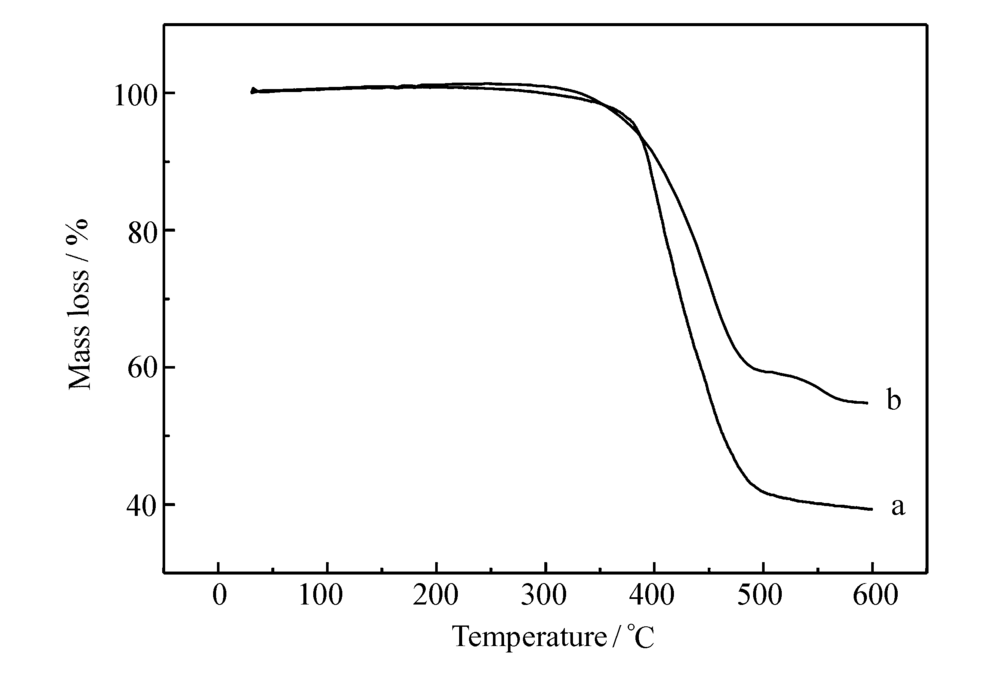 | 图6 P2.5(a)和P10(b)的热失重曲线Fig.6 TGA curves of P2.5(a) and P10(b) |
{kind=link}
合成了以苯基苯并咪唑为环金属配体、吡啶三唑为辅助配体的高效黄绿光铱配合物M1,并作为客体和中心核、以芴-咔唑为主体和臂,通过Suzuki缩聚得到星型聚合物材料。 聚合物的发光光谱大多呈现蓝光和黄光的双极发射,表现主客体能量的部分转移,可通过调节客体单元的比例,来实现聚合物材料的不同发光颜色。 当M1摩尔含量为2.5%时,获得了白光聚合物,随着M1摩尔分数的提高,能量传递更加充分,聚合物的发光颜色逐渐变为黄光。 聚合物的荧光衰减寿命均为微秒级别,符合磷光材料的特点。M1的摩尔分数对聚合物的HOMO能级和LUMO能级影响不大。 聚合物具有较高的热稳定性,能够满足发光材料的要求。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|