

YU Huifa, CHEN Chonglai, WANG Yuejuan, et al. Ozone Decomposition over NiO/Mn3O4 Monolithic Catalysts [J]. Chinese Journal of Applied Chemistry, 36(6): 698-703


将原料Ni(NO3)2·6H2O、Mn3O4粉末和拟薄水铝石用球磨机球磨,以所得的浆料浸渍堇青石,经过焙烧,得到不同比例的NiO/Mn3O4催化剂。 通过催化分解臭氧活性测试发现,在空速为20000 h-1时, 30NiO/Mn3O4(NiO占总质量的30%)催化剂的活性最高,臭氧分解率达到98%,催化剂活性稳定。 当提高空速为40000 h-1,50NiO/Mn3O4(NiO占总质量的50%)催化剂的活性最高,臭氧分解率在90%左右,并且出现失活现象。 通过X射线衍射(XRD)、程序升温(TPR)、X射线光电子能谱分析(XPS)、BET比表面积法等表征,发现Mn3O4和NiO复合催化剂的比表面积大于单一金属氧化物催化剂的比表面积并且在Mn3O4和NiO复合催化剂中Mn3O4与NiO发生电子相互作用。 催化剂中的Mn3O4与NiO的协同催化作用。 使得Mn3O4与NiO混合物催化剂的还原温度降低,分解臭氧(O3)活性提高。
A series of NiO/Mn3O4 monolithic catalysts with different NiO contents was prepared by ball-milling of Ni(NO3)2·6H2O, Mn3O4 and pseudo boehmite precusors via subsequent impregnation with cordierite, followed by calcination. These catalysts were tested for ozone decomposition. It was found that the 30NiO/Mn3O4(mass fraction of NiO in total mass is 30%) catalyst has the highest activity at a space velocity of 20000 h-1, and leads to 98% conversion of ozone, while the catalyst remains stable. When the space velocity is increased to 40000 h-1, the 50NiO/Mn3O4(mass fraction of NiO in total mass is 50%) catalyst gives the highest activity, with a ozone conversion at about 90%. But the catalyst suffers deactivation. Characterizations by X-ray diffraction(XRD), TPR, XPS and BET reveal that the presence of Mn3O4 in the NiO increases the specific surface area of the catalyst, and electronic interaction between Mn3O4 and NiO. Meanwhile, the co-presence of Mn3O4 and NiO in the catalyst results in facile reduction of these oxides. This synergy is believed to be responsible for the enhanced catalytic performance.

臭氧(O3)是氧的同素异型体,在常温常压下是一种不稳定的淡紫色气体。 由于其强氧化性,广泛应用于水和空气的脱臭、杀菌以及废水的脱色、废水中的有机物去除和氰基等有机物的分解[1]。 高浓度的O3会对人体和生物组织造成直接损害。 研究结果表明,当臭氧的浓度为0.24 mg/m3时,呼吸2 h,便使肺活量减少20%;当O3的浓度为0.28 mg/m3时,胸部和鼻子将会产生刺激感。 世界卫生组织已制订了关于臭氧的安全标准:8 h工作环境下允许的最大浓度应低于0.21 mg/m3 [2,3,4,5]。 目前,常用的臭氧尾气处理方法有:热分解法、活性炭吸附法、药剂氧化法、催化分解法、燃烧法和大气稀释排放法等。 各种处理方法各有其优缺点,其中催化分解法由于具有O3分解完全,催化剂可再生重复使用和处理操作简便及无二次污染而更受重视[6]。 催化分解O3的催化剂可大致分为活性炭(AC)、过渡金属氧化物和贵金属3种[7],贵金属(如:钯、铂)或过渡元素金属氧化物(如:Mn、Cu的氧化物)为活性组分,载体则采用 γ-氧化铝、TiO2、SiO2、分子筛、活性炭或以上几种的复合成分。 贵金属价格高昂,应用受限,所以以金属氧化物为主的研究逐渐成为主流[8,9,10,11,12]。 Dhandapani等在臭氧分解研究中得出了MnO2活性最好的结论。 大多数研究者的工作也是以含有MnO2为基础的催化剂上进行改进。 Wu等[13]研究了其他金属及氧化物的催化作用。 为提高分解臭氧的能力,往往需加入贵金属(如银、铂等),它们存在的共同问题是催化剂制作复杂,成本高,耐湿性差,使用寿命短,大多数催化剂在实际工业中很难应用。 本文的研究目的在于研制出一种催化分解臭氧效率高且价廉的NiO、Mn3O4双金属催化剂[14]。
采用Bruker D8 ADVANCE型X射线粉末衍射仪(XRD,德国布鲁克AXS有限公司),室温、空气气氛下对样品进行分析,Cu Kα为激光光源, λ=0.1542 nm,扫描范围为10°~90°;比表面积及孔性质采用Autosorb-1型物理吸附仪(美国Quantachrome公司)进行分析测试,吸附前,催化剂在真空中120 ℃预处理4 h,在-196 ℃下吸附N2;采用ESCALAB 250Xi型X射线光电子能谱仪(XPS)进行元素分析:以单色化Al Kα X射线( hν=1486.6 eV)为激发源,工作电压20 kV,电子结合能以表面沉积碳校正(污染碳的结合能C1 s为284.8 eV),采用XPSPEAK41软件进行分峰拟合;氢气程序升温还原(H2-TPR)实验对催化剂的氧化还原性能进行测试分析,样品先通5%H2+95%N2混合气(30 mL/min)进行还原,装量为25 mg,从40 ℃程序升温至800 ℃(升温速率为20 ℃/min),由配有热导检测器(TCD)的气相色谱仪来记录样品耗氢量。
Mn3O4(质量分数97%,上海麦克林生化科技有限公司,比表面为16 m2/g);拟薄水铝石(AlOOH· nH2O, n=0.08~0.62,纯度99%,青岛山科海泰新材料有限公司);硝酸镍(分析纯,国药集团化学试剂有限公司);堇青石蜂窝陶瓷(38 μm,江西安天高新材料有限公司);臭氧发生器;去离子水(自制);硝酸(分析纯,国药集团化学试剂有限公司)。
1.2.1 催化剂的制备
以堇青石陶瓷为载体,采用浸渍法制备NiO/Mn3O4整体式催化剂。 NiO/Mn3O4负载量45 g/L,即每升堇青石负载45 g的活性组分(NiO和Mn3O4的总质量),15 g拟薄水铝石。 以5NiO/Mn3O4为例:以原料8.8 g Ni(NO3)2·6H2O,42.75 g Mn3O4,15 g拟薄水铝石,配制成混合浆液,采用浸渍法将堇青石蜂窝陶瓷载体浸渍在配制的浆液中,然后,经400 ℃焙烧4 h后得2.25 g NiO,42.75 g Mn3O4,即NiO质量占NiO和Mn3O4总质量的5%,而拟薄水铝石不作考虑,记为5NiO/Mn3O4。 剩余浆液经干燥,在400 ℃焙烧,得到相应的粉末催化剂,用于表征。 采用相同方法制备不同Ni负载量的整体式催化剂,不同Ni负载量的整体式催化剂命名为 xNiO/Mn3O4, x表示NiO/Mn3O4复合氧化物中NiO的质量分数(如 x为5表示催化剂中NiO质量占NiO和Mn3O4总负载量的质量为5%,本文 x选择了0、5、30、50、80和100)。
1.2.3 催化剂的活性评价
常压下在连续流动固定床反应装置上,装填直径19.2 mm,高40 mm的圆柱形整体式催化剂2块,以O3/空气混合气作为反应气,其中空速为20000 h-1,臭氧浓度为70.6 mg/m3,在常温下进行催化分解反应。 每隔10 min检测尾气中臭氧的浓度,用SKY-2000臭氧检测仪测定臭氧。分别取反应前和反应后的气体进行检测,根据反应前和反应后的浓度计算出反应转化率,公式如下:
式中,XO3(%)为臭氧的转化率; ρ(O3)in为反应前O3的质量浓度(mg/m3), ρ(O3)out为反应后O3的质量浓度(mg/m3)。
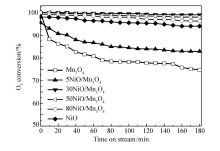
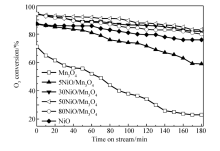
图1和图2为不同空速(20000和40000 h-1),不同O3质量浓度(70.6和21.4 mg/m3)条件下,各种催化剂上O3转化率与反应时间的关系。图1是空速20000 h-1,O3浓度70.6 mg/m3,反应温度20 ℃,催化剂的O3转化率与反应时间的关系。 由图1可见,单组分Mn3O4催化剂活性最低,在3 h后O3的转化率保持在75%左右,催化剂失活非常明显。 单组分NiO催化剂的O3分解活性高于单组分Mn3O4催化剂,在3 h后O3的转化率保持在94%左右。 双组份的NiO/Mn3O4复合催化剂,除了5NiO/Mn3O4催化剂,其它催化剂的O3分解活性明显提高,其中30NiO/Mn3O4的催化剂活性最高,在3 h后O3的转化率仍然保持在98%左右,说明NiO和Mn3O4的复合有利于O3的分解。 由图2可见,50NiO/Mn3O4催化剂的O3分解活性最高,其次分别是30NiO/Mn3O4、80NiO/Mn3O4、NiO、5NiO/Mn3O4和Mn3O4催化剂。 对比图1和图2可知,当空速提高1倍,虽然会造成O3浓度下降30%(70.6至21.4 mg/m3),催化剂上O3转化率明显下降的结果,仍可说明提高空速不利于O3的分解。 催化剂对O3分解的活性顺序,与20000 h-1空速时基本保持一致。 由此可见, 30NiO/Mn3O4、50NiO/Mn3O4和80NiO/Mn3O4催化剂的活性高于单一金属NiO催化剂的活性。 可以推测,Mn3O4和NiO之间可能存在相互作用,有利于催化剂的O3分解。
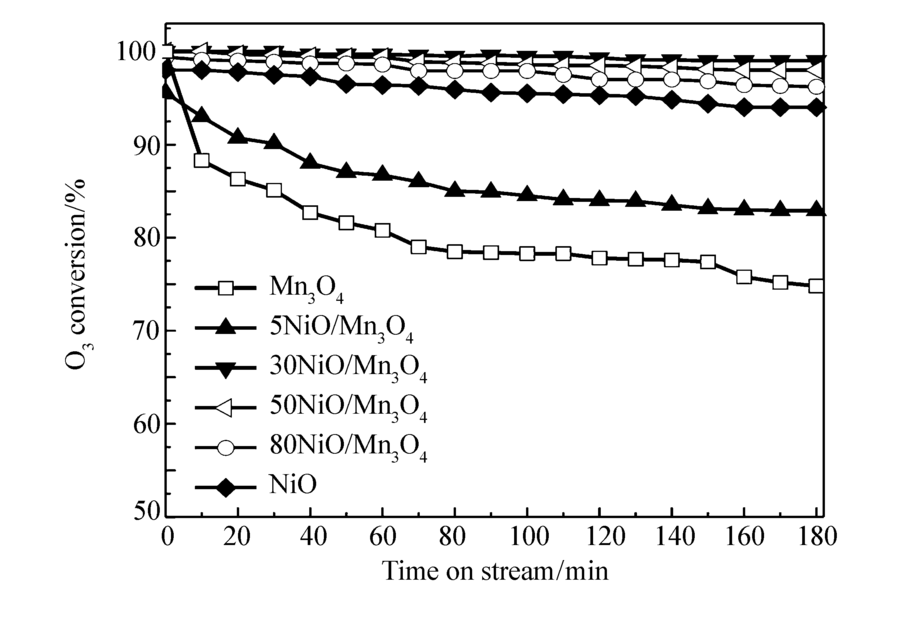 | 图1 NiO/Mn3O4催化剂上O3转化率与反应时间的关系Fig.1 Relationship between O3 conversion and time on stream over NiO/Mn3O4 catalystsReaction conditions:space velocity=20000 h-1, ρ(O3)=70.6 mg/m3, temperature=20 ℃ |
{kind=link}
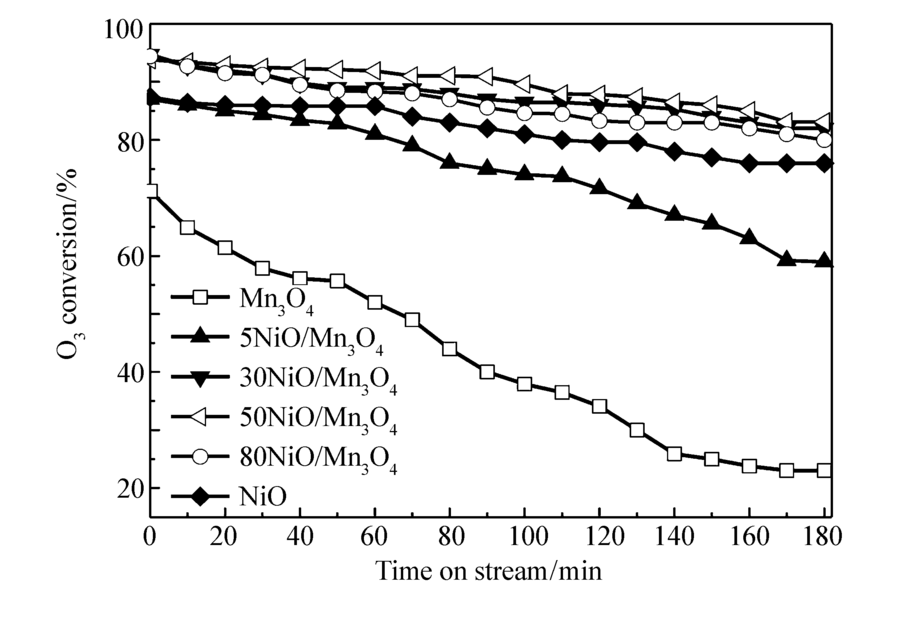 | 图2 NiO/Mn3O4催化剂上O3转化率与反应时间的关系Fig.2 Relationship between O3 conversion and time on stream over NiO/Mn3O4 catalystsReaction conditions:space velocity=40000 h-1, ρ(O3)=21.4 mg/m3, temperature=20 ℃ |
{kind=link}
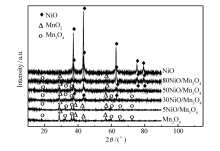
图3是催化剂的XRD谱图。 Mn3O4、5NiO/Mn3O4和30NiO/Mn3O4的催化剂中能观察到Mn3O4(PDF No.24-0734)的特征衍射峰(18°、28.7°、32.3°、36°、59.8°、64.6°和72.3°), β-MnO2(PDF No.24-0735)的特征衍射峰(37.3°、42.8°和56.6°),随着催化剂NiO含量的增加,Mn3O4和 β-MnO2衍射峰强度逐渐降低,然而特征衍射峰的位置没有发生变化,说明Mn3O4和 β-MnO2的结构未发生改变。 30NiO/Mn3O4、50NiO/Mn3O4、80NiO/Mn3O4和NiO的样品中可观察到NiO(PDF No.47-1049)的特征衍射峰(37.2°、43.3°、62.8°、75.4°和79.4°)。 随着催化剂NiO含量的增加,NiO衍射峰强度逐渐增强,然而,在5NiO/Mn3O4的样品中几乎观察不到NiO的特征衍射峰,说明NiO在催化剂表面是高度分散的。
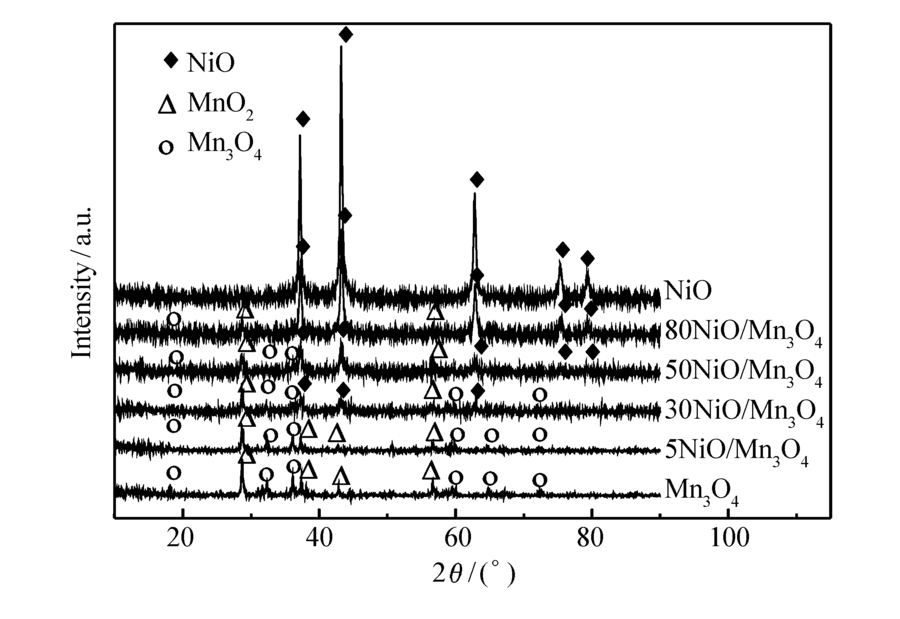 | 图3 NiO/Mn3O4 催化剂的XRD图Fig.3 XRD patterns of NiO/Mn3O4 catalysts |
{kind=link}
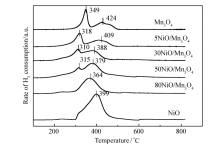
图4是NiO/Mn3O4催化剂的H2-TPR图谱。 由图4可看出,Mn3O4催化剂在349和424 ℃处出现两个还原峰。 从图3的XRD结果,Mn3O4催化剂由 β-MnO2和Mn3O4组成,根据文献[15]将349 ℃处的还原峰归属于MnO2到Mn3O4的还原;424 ℃处的还原峰归属于Mn3O4到MnO的还原。 NiO催化剂在399 ℃处出现了一个非对称的NiO还原峰,可能存在两个还原过程,即Ni2O3到NiO的还原和NiO到Ni的还原[16,17]。 在5NiO/Mn3O4、30NiO/Mn3O4和50NiO/Mn3O4催化剂中观察到两个还原峰,还原峰分别在310~320 ℃之间,370~410 ℃之间,并且还原峰的温度比Mn3O4催化剂明显向低温方向偏移。 80NiO/Mn3O4催化剂中只观察到1个364 ℃的还原峰。 由图4还可以发现,随着NiO含量的增加,低温还原峰(<350 ℃)减小,在80NiO/Mn3O4催化剂中消失,说明低温还原峰是催化剂中 β-MnO2物种的还原,而高温还原峰(>350 ℃)是NiO和Mn3O4的还原,由于NiO和Mn3O4的还原温度比较接近,还原峰重叠在一起了。 从NiO/Mn3O4复合催化剂的还原峰温度低于单一金属氧化物的还原峰温度,推测NiO和Mn3O4发生了相互作用,这种相互作用使得复合氧化物催化剂的O3分解活性提高。
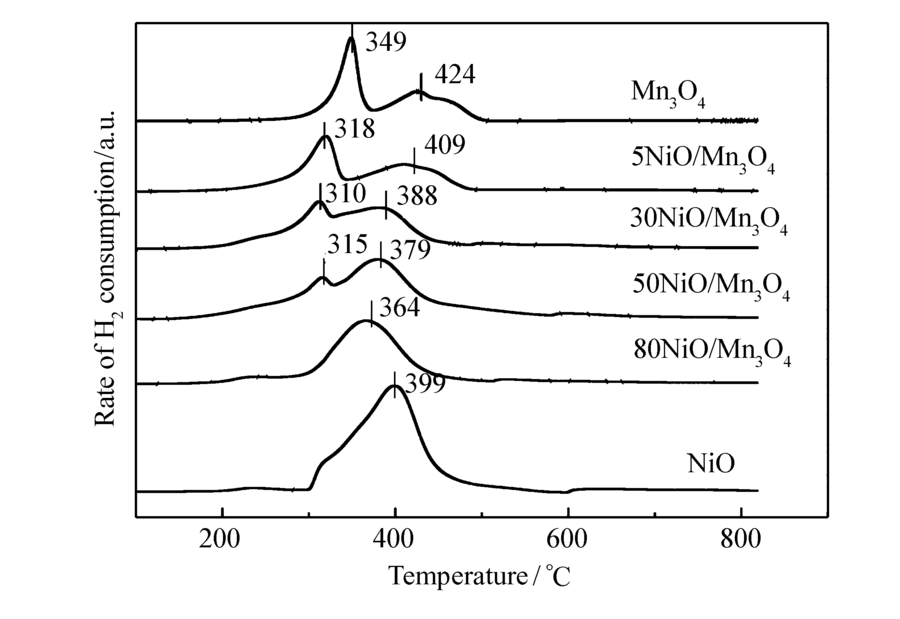 | 图4 NiO/Mn3O4催化剂的TPR图Fig.4 TPR profiles of NiO/Mn3O4 catalysts |
{kind=link}
图5是NiO/Mn3O4催化剂的Mn2 p、Ni2 p和O1 s的XPS图谱。 由图5可见,非对称的Mn2 p3/2可以分为3个峰,电子结合能分别在641.4、642.5和645.2 eV,分别归属Mn2+、Mn3+、Mn4+的Mn2 p3/2能谱。 非对称的Ni2 p3/2通过分峰可分为3个峰,电子结合能在853.7、855.6和861.3 eV分别归属Ni2+、Ni3+以及卫星峰。 然而,催化剂中同一价态元素的电子结合能有所不同,NiO、5NiO/Mn3O4、30NiO/Mn3O4、50NiO/Mn3O4、80NiO/Mn3O4的Ni2+的电子结合能分别为853.7、854.0、854.1、854.2和855.8 eV,Ni3+的电子结合能分别为855.6、855.6、855.9、856.0和855.8 eV。 O1 s可以分为3个峰,电子结合能在529.2、530.8和531.9 eV分别归属于晶格氧,吸附氧,吸附的羟基或水分子氧[17,18]。 由XPS图谱(图5)可知,在NiO/Mn3O4复合催化剂中的结合能与单金属氧化物催化剂的结合能发生了偏移,由此可发现,随着Mn3O4的添加,NiO的Ni2+与Ni3+的电子结合能分别先增加后减小,在50NiO/Mn3O4催化剂中Ni2+与Ni3+的电子结合能发生偏移最大,说明该催化剂中NiO和Mn3O4发生相互作用最强。 这些变化规律与催化剂的催化活性规律是相一致的,说明NiO和Mn3O4发生相互作用越强,催化活性越高,存在共助催化作用[19]。表1是NiO/Mn3O4催化剂的表面元素组成及催化剂的BET比表面积。 由于空白堇青石陶瓷载体的比表面在3~5 m2/g,并且没有催化活性,因此没有列入表格中。 从表1可看出,在Mn3O4、5NiO/Mn3O4、30NiO/Mn3O4、50NiO/Mn3O4、80NiO/Mn3O4催化剂中,Mn2+含量总体随着NiO的增加呈下降趋势,而Mn3+含量总体先增加后减少,但是催化剂表面Mn4+含量随着NiO含量的增加而增加,因此NiO的增加有利于高价态Mn(MnO2)的生成。 NiO/Mn3O4复合催化剂的O3分解活性的提高可能与Mn4+含量的提高有关。 然而对于80NiO/Mn3O4催化剂,Mn4+含量最高,但O3分解活性低于50NiO/Mn3O4催化剂,这说明对于O3分解影响因素是比较复杂的,也可能受到催化剂比表面的影响。
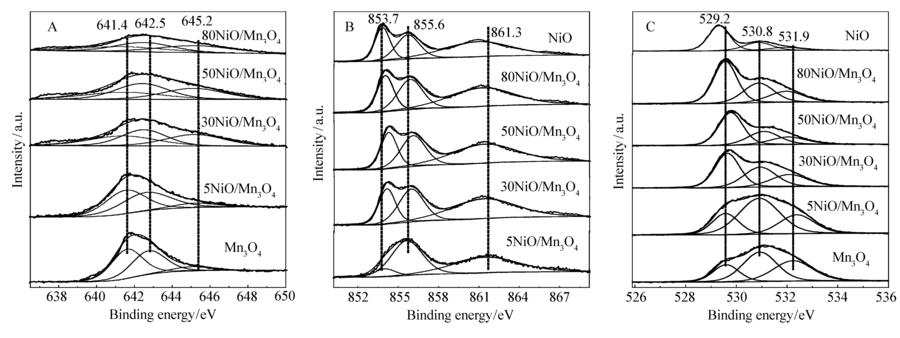 | 图5 NiO/Mn3O4催化剂的Mn2 p(A)、Ni2 p(B)和O1 s(C)的XPS谱图Fig.5 XPS spectra of Mn2 p(A), Ni2 p(B) and O1 s(C) of NiO/Mn3O4 catalysts |
{kind=link}
| 表1 NiO/Mn3O4催化剂的表面元素组成及催化剂的BET比表面 Table 1 Surface element composition and BET specific surface area of NiO/Mn3O4 catalysts |
根据催化剂的O3分解的活性, Mn3O4催化剂的催化分解臭氧活性最低,随着添加的NiO含量的增加,催化剂催化活性也是不断提升的。 但是,NiO催化剂的活性反而比30NiO/Mn3O4、50NiO/Mn3O4、80NiO/Mn3O4催化剂活性低,说明共助催化作用。 因此,可提出该催化反应的机理如式(1)-(3)所示:
O3 + * → O* + O2 (1)
O3 + O* → O2* + O2 (2)
O2* → O2+ * (3)
O3催化分解反应机理可能是电子从活性位(低价态的Ni、Mn离子)传输到O3分子,形成了高价态的Ni、Mn离子和原子氧并且释放出氧气。 随后带有原子氧的高价态Ni、Mn离子与臭氧分子结合,形成另外一种带有分子氧的高价态Ni、Mn离子并释放出氧气分子。 最后分子氧的高价态离子释放出氧气分子并形成低价态Ni、Mn离子。 这样形成了整个催化循环过程[20,21]。 可见,NiO/Mn3O4复合催化剂中,NiO和Mn3O4之间的相互作用,Mn价态的变化可能促进了O3的分解。
通过对NiO/Mn3O4复合催化剂的O3分解性能的比较,发现NiO/Mn3O4复合催化剂具有较高的O3分解性能,以30NiO/Mn3O4和50NiO/Mn3O4复合催化剂的活性最高,在室温、空速20000 h-1条件下,O3分解的转化率达到98%左右,而且催化剂活性比较稳定。 在NiO含量较低时,NiO物种以分散状态存在,随后随着NiO量的增加,NiO晶粒增大,NiO/Mn3O4复合催化剂的还原温度较单一金属氧化物催化剂低,分解臭氧的活性高于单一金属氧化物催化剂。 在NiO/Mn3O4复合催化剂中,即Mn3O4基催化剂通过NiO的改性,使得NiO和Mn3O4的电子发生相互作用和协同作用,提高了催化剂的臭氧分解活性。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|