
DONG Zhaoxia, SONG Tongyang, LU Jiqing, et al. Gas Phase Dehydrochlorination of 1,1,2-Trichloroethane over Cr2O3 Catalysts [J]. Chinese Journal of Applied Chemistry, 36(5): 515-523


共同通讯联系人:谢冠群,高级工程师; Tel:0579-82291381; E-mail:gqxie@zjnu.cn; 研究方向:催化化学
采用沉淀法制备了不同焙烧温度的Cr2O3催化剂,用于1,1,2-三氯乙烷(TCE)气相脱氯化氢制备二氯乙烯的反应。 采用X射线衍射(XRD)、氢气程序升温还原(H2-TPR)、氨气程序升温脱附(NH3-TPD)、X射线光电子能谱(XPS)表征手段,研究了Cr2O3催化剂气相催化裂解TCE脱氯化氢反应及其反应机理。 结果表明,Cr2O3催化剂上TCE气相脱氯化氢反应的转化率随着催化剂焙烧温度的升高逐渐降低,然而顺-1,2-二氯乙烯( cis-DCE)的选择性先增大后减小。 400 ℃焙烧的Cr2O3催化剂催化性能最好,TCE转化率为70.8%,顺-1,2-二氯乙烯的选择性为90.0%。 然而,催化剂的单位面积反应速率随着焙烧温度升高先提高后下降,400 ℃焙烧催化剂的单位面积反应速率为0.801×10-2 μmol/(s·m2)。 催化剂的单位面积反应速率和顺-1,2-二氯乙烯( cis-DCE)的选择性与催化剂表面Cr2O3物种具有很好的对应关系,表明催化剂表面Cr2O3物种有利于脱氯化氢反应。 以酸中心为活性中心计算得到的转换频率(TOF)变化趋势与单位面积反应速率相一致,400 ℃焙烧的催化剂的TOF为2.82×10-5 s-1,表明Cr2O3催化剂Cr物种合适的平均价态(~3.20)有利于脱氯化氢反应。
Co-corresponding author:XIE Guanqun, senior engineer; Tel:0579-82291381; E-mail:gqxie@zjnu.cn; Research interests:catalytical chemistry
A series of Cr2O3 catalysts was prepared by a precipitation method and tested for the gas phase dehydrochlorination of 1,1,2-trichloroethane(TCE) to synthesize cis-1,2-dichloroethylene( cis-DCE). X-ray diffraction(XRD), hydrogen temperature-programmed reduction(H2-TPR), ammonia temperature-programmed desorption(NH3-TPD) and X-ray photoelectron spectroscopy(XPS) were used to study the dehydrochlorination of TCE on Cr2O3 catalyst and its reaction mechanism. It is found that the conversion ratio of TCE on the catalysts decreases with the increase of the calcination temperature, while the selectivity to cis-DCE first increases and then decreases. The best performance is obtained on the catalyst calcined at 400 ℃, with a TCE conversion ratio of 70.8% and a cis-DCE selectivity of 90.0%. In addition, the areal specific reaction rate also first increases and then decreases with the increase of the calcination temperature, with the highest value being obtained on the catalysts calcined at 400 ℃(0.801×10-2 μmol/(s·m2). The catalytic behaviors of the catalysts are well related to their surface Cr2O3 species. Turnover frequencies(TOFs) calculated based on the surface acidity and the highest value being obtained on the catalysts calcined at 400 ℃(2.82×10-5 s-1) show that the oxidation states of the surface Cr species are important for the reaction and the Cr species with an average valence of 3.2 are appropriate for the reaction.



氯代有机物种类繁多,是重要的化工原料,但由于不合理的废水排放等原因,对生态系统和地下水安全造成严重威胁[1],因此如何利用氯代烃类物质是一个重要课题。 气相催化裂解氯代烷烃脱氯化氢有经济高效,环境污染小的优点,越来越受到学术界和工业界的关注。 Mukhopadhyay等[2]采用活性炭催化脱氯化氢,反应的转化率为100%和选择性为96%。 Wang等[3]对活性炭催化剂的稳定性问题进行了考察,发现活性炭用盐酸或者氢氟酸去矿化,用硝酸、过氧化氢、氧气氧化处理过后稳定性显著提高。 Li等[4]考察了纳米TiO2上催化1,1,2,2-四氯乙烷脱氯化氢反应:晶相和暴露面的作用,发现纳米TiO2的结晶相和暴露面影响了其吸附能力和表面催化活性,进而影响反应的催化效率。
目前,对于气相催化1,1,2-三氯乙烷(TCE)脱氯化氢反应的研究非常广泛,其主要产物为偏二氯乙烯(VDC),顺式1,2-二氯乙烯( cis-DCE)[5,6,7]和反式1,2-二氯乙烯( trans-DCE)[8]。 通过脱氯方法不仅可以解决氯代烷烃对环境的污染问题,而且可对其产物进行选择性的生产以发挥其良好的应用价值,如TCE脱氯化氢生成的顺-1,2-二氯乙烯是重要的化工基础原料,常用作杀菌剂、麻醉剂和冷冻剂,也可以用作涂料、树脂、蜡、橡胶和醋酸纤维的溶剂[9]。
碱金属、过渡金属负载型催化剂常用于气相催化裂解TCE反应,靳燕霞等[10]发现,400 ℃下在CsNO3/SiO2催化剂上裂解TCE脱氯反应100 h,TCE转化率和VDC选择性分别能稳定在达到98%和78%。 Tang等[9]的研究表明Mg负载量为10%的Mg/SiO2催化剂在气相裂解TCE反应中催化活性最好,TCE转化率和 cis-DCE选择性均达到90%以上,且反应中形成的Mg(OH)Cl物种为活性中心。 胡益浩等[11]研究了SiO2负载过渡金属催化剂(M/SiO2)的气相裂解TCE脱氯反应,发现Zn/SiO2催化性能最好,TCE转化率达到98%, cis-DCE的选择性为82%。 Song等[12]考察了SiO2负载碱金属和过渡金属催化剂的酸碱性对TCE脱氯化氢性能的影响,发现催化剂的表面碱性有利于VDC生成,酸性有利于生成 cis-DCE。
然而,对于气相催化裂解TCE工艺,催化剂容易失活是制约工业化的主要因素。 因此,研究催化剂的失活,提高催化剂的稳定性对于气相催化裂解TCE工艺非常重要。 研究发现催化剂表面强酸中心是导致催化剂积炭和失活的主要原因[11]。 也有研究认为CsNO3/SiO2催化剂失活的主要原因是含氯反应产物在低温反应时难以从催化剂表面脱附[10]。 铬(Cr)基催化剂在许多催化反应中表现出高活性和良好的稳定性,广泛用于含氟烷烃脱氟化氢反应并进行了深入的研究。 Wang等[13]采用铬基催化剂用于气相氟化2-氯-1,1,1-三氟乙烷合成1,1,1,2-四氟乙烷,发现导致CrO3/Cr2O3催化剂失活的原因是催化剂经HF反应后生成了CrO xF y活性物种,在反应中转化成稳定但无活性的CrF3。 Han等[14]研究了氟化的Cr2O3催化剂上裂解1,1-二氟乙烷脱氟化氢制备氟乙烯的反应,发现制备的六方棱柱的纳米Cr2O3有更高的比表面积、更多的L酸且在反应中形成的CrO xF y提升了反应的活性和稳定性。 Lim等[15]考察了原位氟化的氧化铬催化剂上气相催化1,1,1,2,3-五氟丙烷脱氟化氢制备2,3,3,3-四氟丙烯的反应,得到较好的结果。 Luo等[16]考察了氟化的NiO/Cr2O3催化剂上气相催化1,1,1,3,3-五氟丙烷脱氟化氢制备1,3,3,3-四氟丙烯的反应,指出反应中形成的NiF2提供新的酸性位点较多且表面酸密度较小,是活性高且减少积碳的原因。
考虑到脱氟化氢和脱氯化氢存在的共性,进行了Cr2O3催化剂上1,1,2-三氯乙烷气相脱HCl反应的研究。 本文用沉淀法制备Cr(OH)3催化剂,经高温焙烧得到Cr2O3催化剂,通过对催化剂焙烧温度等因素的实验探究,结合X射线衍射(XRD)、氢气程序升温还原(H2-TPR)、氨气程序升温脱附(NH3-TPD)、X射线光电子能谱(XPS)等表征手段,详细研究了Cr2O3催化剂气相催化裂解TCE脱氯化氢反应及其反应机理。
Bruker D8 ADVANCE型X射线粉末衍射仪(XRD,德国Bruker公司),Autosorb-Ⅰ型物理吸附仪(BET,美国Quantachrome公司),ESCALAB 250Xi型X射线光电子能谱仪(XPS,美国Thermo Fisher Scientific公司),火焰粒子检测器(FID)的气相色谱(GC, 美国Agilent 6850 DB-1,色谱柱30 m×0.32 mm)。 TCE(纯度99.8%)购自上海阿拉丁生化科技股份有限公司,Cr(NO3)3·9H2O,浓NH3·H2O均购自国药集团化学试剂有限公司,均为分析纯试剂。
采用沉淀法制备Cr(OH)3催化剂,并高温焙烧得到Cr2O3催化剂,具体如下:称取25.000 g Cr(NO3)3·9H2O溶于150 mL去离子水,得到深紫色溶液;量取25 mL浓NH3·H2O (国药集团化学试剂有限公司)溶于150 mL 去离子水;将Cr(NO3)3溶液逐滴滴入NH3·H2O溶液中,搅拌状态下至溶液pH=9;静置,抽滤,之后将样品于80 ℃烘箱烘干12 h;研磨,得到Cr(OH)3前驱物,然后分别在300、400、500、600和700 ℃下高温焙烧4 h(程序升温,20 ℃/min)得到Cr2O3催化剂,以Cr2O3-300、Cr2O3-400、Cr2O3-500、Cr2O3-600和Cr2O3-700分别表示不同焙烧温度的催化剂。
对样品XRD分析采用Cu Kα为激光光源, 波长0.1542 nm。 扫描范围为10°~90°,样品测试在室温及空气气氛中进行。
比表面积及孔性质采用BET进行分析测试,吸附前,催化剂在真空中120 ℃预处理4 h,在-196 ℃下吸附N2。
XPS检测:以单色化Al Kα X射线( hν=1486.6 eV)为激发源,工作电压20 kV。 电子结合能以表面沉积碳校正(污染碳的结合能C1 s为284.8 eV)。 采用XPSPEAK41软件进行分峰拟合。
采用H2-TPR实验对催化剂的氧化还原性能进行测试分析。 样品先通5% H2+95% N2混合气(30 mL/min)进行还原,装量为25 mg。 从40 ℃程序升温至800 ℃(升温速率为20 ℃/min),由配有热导检测器(TCD)的GC来记录样品耗氢量,H2实际消耗量用CuO外标进行标定。
催化剂表面酸量和强度分布由NH3-TPD实验装置(自制)进行分析,具体过程如下:取0.2 g Cr2O3催化剂于石英管中,在 N2气气氛400℃预处理0.5 h,流速为20 mL/min ,之后50 ℃吸附NH30.5 h(20 mL/min),然后在100℃下通N2气(20 mL/min)吹扫0.5 h,以除去催化剂表面物理吸附的NH3。 之后从60 ℃升温至800 ℃(20 ℃/min),用TCD来检测和记录NH3脱附信号。
采用自制固定床反应装置,石英反应管内径为8 mm。催化剂活性测试过程如下:取0.10 g催化剂(125~150 μm)置于反应管中,热电偶插入石英管内催化剂的中心位置以监测和控制温度。 Cr2O3催化剂在N2气气氛(10 mL/min)中以20 ℃/min的升温速率,从室温升至400 ℃预处理0.5 h(300 ℃焙烧的催化剂预处理温度为300 ℃),之后降至反应温度250 ℃,进行气相裂解TCE脱氯化氢反应。 N2通过冰水浴冷却的装有TCE饱和器中,N2将TCE带入催化剂床层,流速为10 mL/min,对应的空速为3000/h。 用配有氢火焰检测器(FID)的GC分析反应产物,根据色谱峰面积和校正因子计算反应转化率和产物选择性。
图1是不同焙烧温度下制备的Cr2O3催化剂和催化TCE气相脱附HCl反应后催化剂的XRD图谱,从图1A中可以看出,300 ℃焙烧下制备的Cr2O3催化剂为无定型状态;400~700 ℃焙烧的催化剂为单一Cr2O3(PDF#38-1479) 物相,可以看出随着焙烧温度提高,Cr2O3衍射峰强度明显增大。 从图1B可见,300 ℃焙烧催化剂经过催化反应后出现了晶相Cr2O3衍射峰,说明反应过程促使了无定型CrO x向晶相Cr2O3的转化。然而400~700 ℃焙烧催化剂在反应前后物相没有改变,反应后催化剂的衍射峰强度和反应前的衍射峰基本相同,表明经过反应后催化剂物相未发生明显的变化。表1为不同焙烧温度下制备的Cr2O3催化剂的比表面积和Cr2O3的平均粒径尺寸。 从表1可见,随着焙烧温度的升高,Cr2O3平均粒径增大;催化剂的比表面下降,这是由于焙烧温度的升高,催化剂发生了晶化,导致催化剂Cr2O3的晶粒变大,比表面积减小。
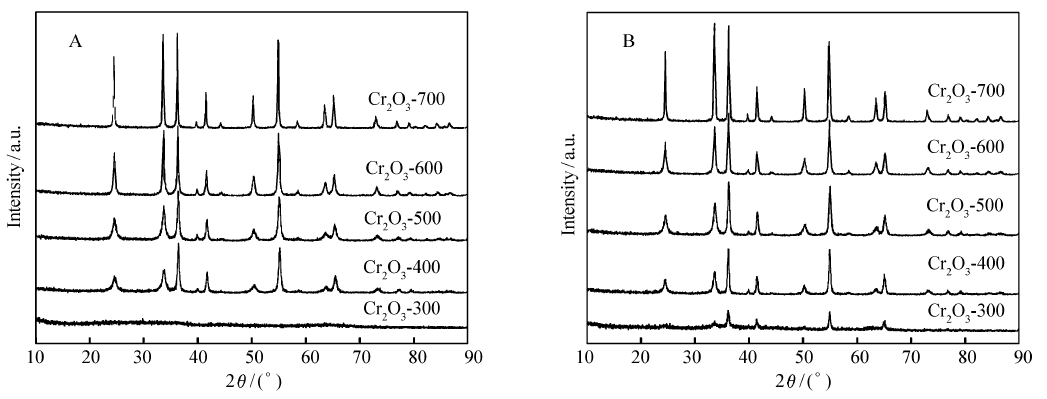 | 图1 不同焙烧温度下制备的Cr2O3催化剂催化反应前(A)后(B)XRD图谱Fig.1 XRD patterns of Cr2O3 catalysts calcined at different temperatures before(A) and after(B) catalytic reaction |
{kind=link}
| 表1 不同焙烧温度下制备的Cr2O3催化剂平均粒径尺寸与比表面积 Table 1 The average crystalline size and specific surface area of Cr2O3 catalysts calcined at different temperatures |
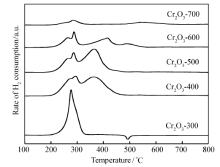
图2是不同焙烧温度Cr2O3催化剂的H2-TPR图,从图2中可以看出,不同温度下焙烧的Cr2O3均出现了两个明显的还原峰。 根据文献[17,18,19],H2-TPR温度低于800 ℃时Cr2O3不能被还原,因此300~400 ℃处的两个还原峰可归属于高价态Cr物种(如CrO3)的还原[18,20,21,22]。 对于Cr2O3-300催化剂在480 ℃附近出现倒峰,这可能是由于催化剂表面存在H2化学吸附,当温度升高时发生脱附产生的,这种现象在我们早期的研究中也发现过[23,24]。 然而,400~700 ℃焙烧的Cr2O3催化剂没有观察到480 ℃附近的倒峰,因此480 ℃附近的倒峰可能与无定型结构Cr2O3有关。 随着催化剂焙烧温度的提高,H2-TPR峰面积逐渐减小。 以CuO为外标,计算催化剂的耗氢量为,Cr2O3-300、Cr2O3-400、Cr2O3-500、Cr2O3-600、Cr2O3-700分别为9.3、1.6、1.5、0.88和0.31 mmol/gcat.。 前期研究表明[18,19],Cr2O3在实验条件下不能继续还原,因此TPR峰是由高价态的氧化铬还原为Cr2O3产生的。 假设催化剂中Cr的价态为(3+ x)而经过TPR实验后Cr的价态为3,那么通过耗氢量可以计算出 x,也就知道了催化剂中Cr的平均价态(3+ x)。 从TPR实验的定量计算可得到Cr2O3-300、Cr2O3-400、Cr2O3-500、Cr2O3-600和Cr2O3-700的Cr平均价态为4.66、3.25、3.23、3.14及3.05。 由此可见,焙烧温度为300 ℃的Cr2O3催化剂中存在高价铬,高价态Cr主要为Cr
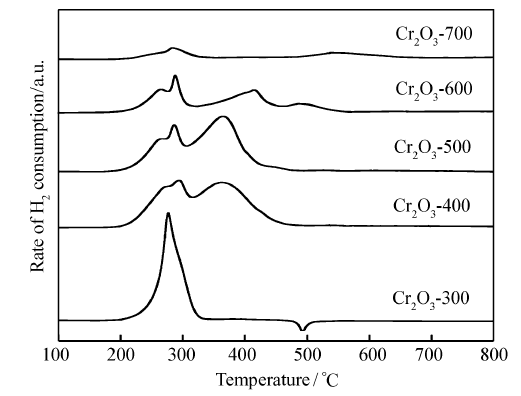 | 图2 不同焙烧温度下制备的Cr2O3催化剂的H2-TPR图Fig.2 H2-TPR profiles of Cr2O3catalysts calcined at different temperature |
{kind=link}
图3是不同焙烧温度下制备的Cr2O3催化剂中Cr2 p的XPS谱图。 从图3A的不同焙烧温度Cr2O3催化剂中Cr2 p的XPS谱图可以看出,Cr2O3催化剂的Cr2 p3/2分裂为3个峰,分别在575.9、577.1和579.1 eV左右。 575.9 eV的峰归属于Cr(OH
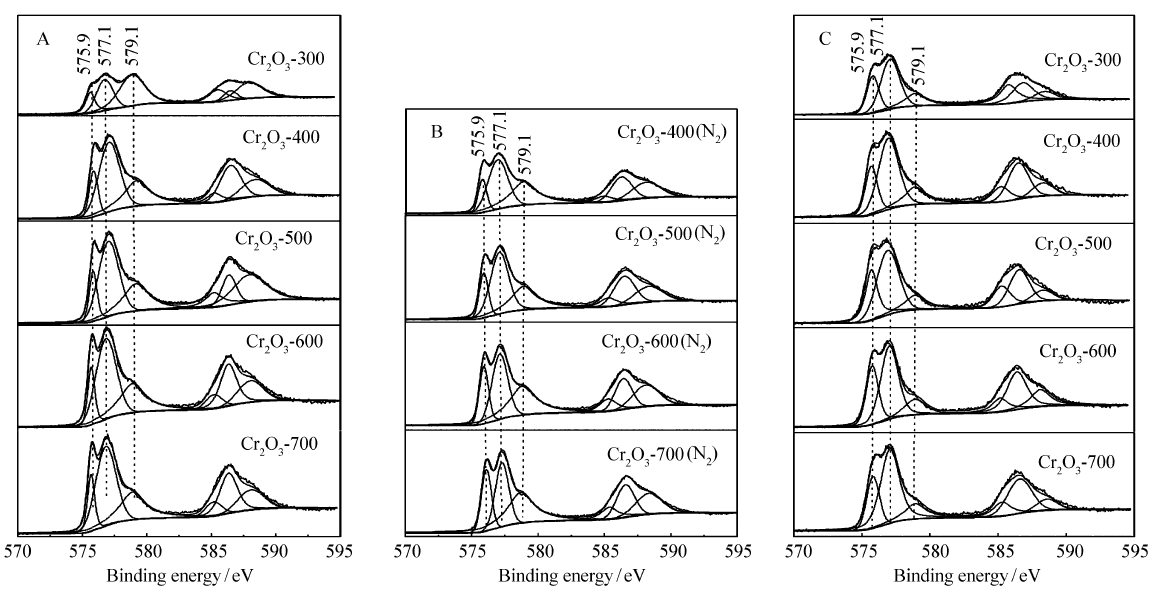 | 图3 不同焙烧温度下制备的Cr2O3(A)、经N2原位预处理(B)或催化反应后(C)催化剂中Cr2 p的XPS谱图Fig.3 Cr2 p XPS spectra of Cr2O3 calcined at different temperatures before(A) and after N2 in situ pretreatment(B) or catalytic reaction(C) |
{kind=link}
| 表2 不同焙烧温度下制备的催化剂表面Cr物种摩尔分数 Table 2 Molar fraction of surface Cr species in Cr2O3 catalysts calcined at different temperatures a |
| 表3 不同焙烧温度催化剂催化反应后的表面Cr物种摩尔分数 Table 3 Molar fraction of surface Cr species in Cr2O3 catalysts calcined at different temperatures after catalytic reaction a |
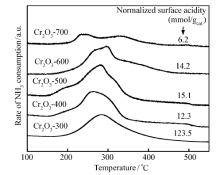
图4是不同焙烧温度下制备的Cr2O3催化剂的NH3-TPD图,从图4可以看出,随着焙烧温度的升高,Cr2O3催化剂的NH3脱附峰位置逐渐向低温方向移动,酸强度逐渐减弱,变化范围在250~285 ℃内,属于中强酸[30]。 根据NH3-TPD脱附物的定量分析,得到催化剂的酸中心数目(图4内),发现随着焙烧温度从300 ℃提高到700 ℃,酸中心浓度从123.5下降到6.2 mmol/gcat。
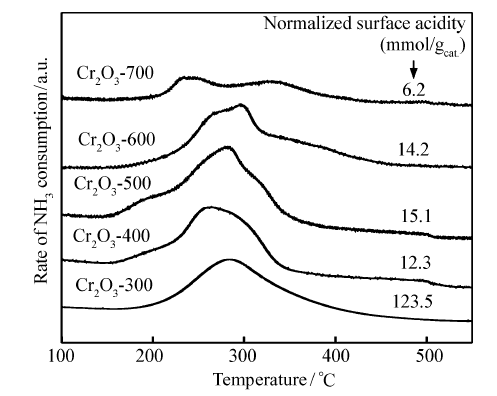 | 图4 不同焙烧温度下制备的Cr2O3催化剂的NH3-TPDFig.4 NH3-TPD profiles of the Cr2O3 catalysts calcined at different temperatures labeled by the normalized surface acidity |
{kind=link}
表4是不同焙烧温度下制备的Cr2O3催化剂上裂解TCE的脱氯性能。 从表4中可见,当焙烧温度为300 ℃时,反应转化率最高达到99.9%,随着焙烧温度的升高,反应的转化率逐渐下降。 结合表1,由于焙烧温度升高时,催化剂Cr2O3的晶粒变大,比表面积减小,反应的转化率也随之逐渐减小,催化活性下降。 顺式 cis-DCE的选择性变化趋势则有所不同,焙烧温度在400、500和600 ℃时 cis-DCE的选择性较高,基本保持在90.0%左右。 其原因可能是400 ℃以上焙烧制备的催化剂中Cr2O3含量的增加,有利于 cis-DCE的生成。 对于脱氯反应,催化剂表面酸性中心是反应的活性中心[11],而Cr2O3催化剂属于中强酸,有利于TCE脱氯化氢生成 cis-DCE[12]。 对于焙烧温度为700 ℃时转化率只有7.0%,催化活性明显下降,这可能与其他因素有关。
| 表4 不同焙烧温度Cr2O3催化剂的裂解三氯乙烷脱氯性能 Table 4 Catalytic performance of Cr2O3 catalysts calcined at different temperatures for dehydrochlorination of TCE |
图5是Cr2O3-400和Cr2O3-300催化剂上脱氯反应的稳定性。 从图5A可见,Cr2O3-400催化剂的脱氯反应反应10 h内活性保持稳定。 通过上述对比得出焙烧温度为400 ℃时,反应温度为250 ℃,TCE的转化率达到70.8%, cis-DCE的选择性达到90.0%,催化性能最好,因此400 ℃为最佳的焙烧温度。 对比表2和表3的新制备的催化剂和反应后催化剂的Cr6+/Cr3+摩尔比可以看出,反应后Cr6+/Cr3+摩尔值比新制备的催化剂下降了一半,但是未观察到催化剂反应性能的变化,可见Cr6+/Cr3+摩尔比不是决定催化剂活性的主要因素。图5B是Cr2O3-300的稳定性实验(催化剂用量为0.05 g),从图5中可以发现,经10 h反应后的转化率从80.0%下降至75.0%,考虑到Cr6+/Cr3+摩尔比不是决定催化剂活性的主要因素,那么Cr2O3-300催化剂活性下降应该归因于催化剂由无定形向晶型转化造成的。
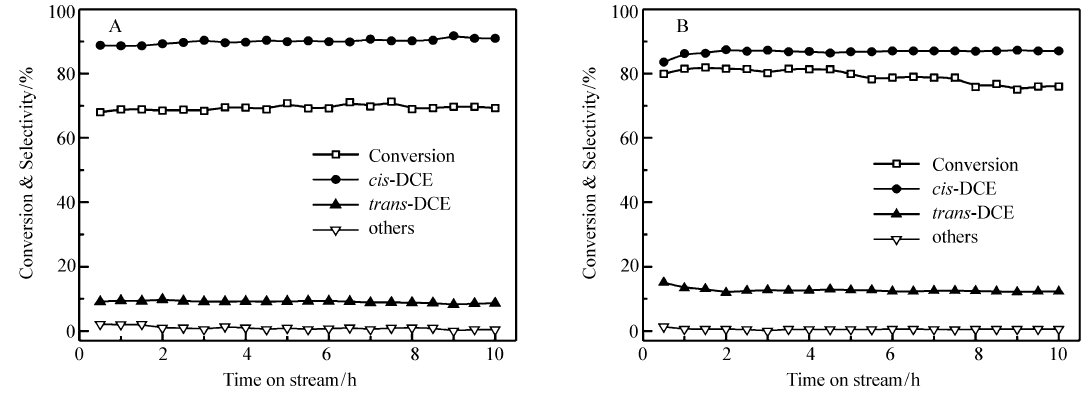 | 图5 Cr2O3-400(A)和Cr2O3-300(B)催化剂裂解三氯乙烷脱氯的反应稳定性Fig.5 Stability of Cr2O3-400(A) and Cr2O3-300 catalyst(B) for dehydrochlorination of TCE Reaction condition:amount of catalyst:A.0.10 g; B.0.05 g; reaction temperature:250 ℃ |
{kind=link}
表5是不同焙烧温度下制备的Cr2O3催化剂的面积比速率和转换频率(TOF)值,TOF值是以表面酸中心为脱氯化氢的活性中心来计算的。 由于表3中Cr2O3-300催化剂的脱氯反应的转化率接近100%,计算TOF会产生巨大的误差,因此,为了提高数据的准确性,将催化剂用量减半为0.05 g(即空速提高1倍,其它反应条件相同)。 这时催化剂的反应转化率为78.5%,以此来计算Cr2O3-300的面积比速率和TOF。 从表5中可见,随着化剂制备的焙烧温度升高,该脱氯反应的面积比速率和TOF值均呈先增加后减小的趋势。 Cr2O3-300催化剂由于比表面积大和酸性位增多,使得单位面积反应速率和TOF下降。 然而700 ℃焙烧的样品虽然表面物种组成与400 ℃相近,但面积比速率与TOF下降非常明显,这意味着催化剂体相Cr的平均价态也影响着催化性能,平均价态在~3.2比较合适,有利于催化剂活性的提高。
| 表5 不同焙烧温度下制备的Cr2O3催化剂的催化反应面积比速率和TOF值 Table 5 Area specific reaction rate and TOF of Cr2O3 calcined at different temperatures |
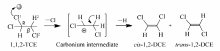
对于该脱氯反应来说,由于TCE分子式中有两类Cl原子(连在 α-C上和 β-C上),产物选择性的差异在于脱除的Cl原子不同。 我们在前期工作中[12]发现 α-C上的Cl原子电荷密度-0.088比 β-C上的Cl原子电荷密度0.041更负,这说明 α-C上的Cl原子更优先与催化剂的酸性位相互作用。 Cr2O3催化剂属于中强酸性催化剂,其表面的酸中心为活性位点,进一步证明了E1机理(Scheme 1):Cr2O3的酸性位与 α-C上的Cl原子相互作用后脱去,形成 α碳正离子中间体,之后 β-C上的H原子离去,形成1.2-二氯乙烯( cis-DCE和 trans-DCE)。
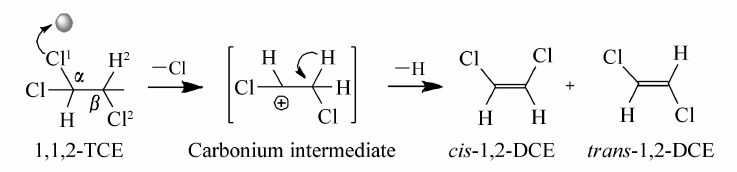 | Scheme 1 E1 reaction mechanism on Cr2O3 catalyst for dehydrochlorination of TCE |
{kind=link}
本文考察了不同焙烧温度下制备的Cr2O3催化剂气相1,1,2-三氯乙烷(TCE)脱HCl性能,结果表明:1)300 ℃焙烧制备的Cr2O3催化剂中铬的平均价态为4.66,600 ℃焙烧的则下降为3.05。 催化剂表面以Cr(OH)3、Cr2O3和CrO3物种形式存在,300 ℃焙烧制备的催化剂表面以CrO3物种为主,400~700 ℃焙烧催化剂表面以Cr2O3物种为主。 2)Cr2O3催化剂上TCE气相脱氯化氢反应的转化率随着制备催化剂焙烧温度的升高逐渐减小,然而顺-1,2-二氯乙烯( cis-DCE)的选择性先增大后减小。 400 ℃焙烧的Cr2O3催化剂的TCE转化率为70.8 %, cis-DCE 的选择性为90.0%;结果表明,单位面积反应速率为0.801×10-2 μmol/(s·m2),转换频率(TOF)为2.82×10-5 s-1,催化剂中Cr的平均价态在~3.2,催化剂的脱氯反应活性最高。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|