
YE Lin, ZHANG Shaofeng, MIN Jiakang, et al. Poly( n-butyl methacrylate)/Polystyrene Blend Compatibilized by the Diels-Alder Reaction of Furan/Maleimide [J]. Chinese Journal of Applied Chemistry, 36(4): 451-458


共同通讯联系人:马丽,副研究员; Tel:0431-85262683; Fax:0431-85262827; E-mail:mali@ciac.ac.cn; 研究方向:高分子的可控合成
利用呋喃和马来酰亚胺之间的可逆Diels-Alder反应,对不相容的聚甲基丙烯酸正丁酯(Poly( n-butyl methacrylate),PBMA)/聚苯乙烯(Polystyrene,PS)合金进行了增容研究。 首先,通过原子转移自由基聚合(ATRP)共聚合成侧基含呋喃基团的聚甲基丙烯酸正丁酯(P(BMA- co-FMA)),并通过后反应改性合成侧基含马来酰亚胺基团的聚苯乙烯(MPS),然后利用呋喃和马来酰亚胺基团之间的Diels-Alder反应促使该共混物中两种高分子的相容性得到明显改善,本实验通过核磁共振波谱仪(NMR)表征验证了功能化高分子及其共混物中的Diels-Alder反应,并通过透射电子显微镜观测相分离结构变化和差示扫描量热法测定相变温度证明了相容性的明显改善。 共混物可以进行热塑性加工,且其相形态和力学性能可以通过控制反应时间予以调节,三点弯曲试验发现随着反应时间的延长共混物逐渐由韧性材料向脆性材料转变。
The compatibilization of immiscible poly( n-butyl methacrylate)(PBMA)/polystyrene(PS) blends was realized by the reaction of furan and maleimide groups. The furan-functionalized poly( n-butyl methacrylate)(P(BMA- co-FMA)) and maleimide-functionalized polystyrene(MPS) was synthesized by copolymerization and post-polymerization modification, respectively. The Diels-Alder reaction of the functionalized polymers and their blend were confirmed by nuclear magnetic resonance spectroscopy(NMR). The reaction of furan and maleimide dramatically promoted the compatibility of two components, confirmed by a homogeneous microstructure evidenced by transmission electron microscopy(TEM) observation and by the presence of a single glass transition temperature in the (P(BMA- co-FMA)/MPS blends. The properties of the blend materials could be controlled conveniently by adjusting the reaction time, and the blends transformed from tough material to brittle material gradually as the reaction processed, which was confirmed by the three point bending test.



高分子合金是由两种或多种高分子通过共混得到的高分子材料。 事实上,与开发新的种类的高分子化合物相比,利用已有的高分子进行共混得到新的高分子材料选择范围更广,是制备新型高分子材料最有效且经济的方法[1]。 根据其各组分的相容性,高分子合金可分为不相容合金、部分相容合金和完全相容合金[2]。 实际应用中,多数共混高分子,例如聚苯乙烯(PS)和聚甲基丙烯酸甲酯(PMMA),属于不相容合金,这种不相容性使共混物存在相分离结构并导致相界面之间作用力较弱,进一步对高分子合金材料的性能产生负面影响。 为解决此问题,研究人员探索了多种方法以控制高分子合金中的相分离和增强合金中的相界面作用。 其中,以下2种方法最为常用[3]:1)添加增容剂,增容剂可以是嵌段/接枝共聚物、纳米粒子等;2)通过化学改性的方法在高分子合金的组分高分子之间引入特定的相互作用或化学反应以增加其相容性。 前者的缺点在于高分子的高黏度导致增容剂很难充分分散到相界面上,可能在某一相中形成胶束或者团聚体。 因此,这种方法增容的聚合物共混物始终是多相体系,很难获得相容的共混物[4]。 在第1种方法中,通过两种高分子之间的特定的化学或者物理作用,在两相界面上原位形成嵌段或者接枝共聚物起到增容剂的作用。 若采用溶液共混方法制备聚合物合金,第2种增容方法甚至可以获得完全相容的聚合物合金[5]。 为确保反应发生在不同的高分子之间,不同的高分子中需要引入不同的功能基团。 目前,已经有多种共价和非共价的相互作用被应用于增容高分子合金体系中,例如,酯交换反应[6]、氢键作用[7]和主客体相互作用[8]。
PS和聚甲基丙烯酸正丁酯(Poly( n-butyl methacrylate),PBMA)是两种典型的不相容高分子。 目前,有多种不同相互作用被应用于PS/PBMA共混体系的增容研究,其中大部分是非共价相互作用。 例如,主客体相互作用[8]、氢键作用[7]以及一些其它基团自身的亲和作用[9]。 呋喃/马来酰亚胺可逆共价连接应用于PS/PBMA共混体系的增容未见报道。
本工作研究了呋喃/马来酰亚胺之间的Diels-Alder(DA)反应对于PS/PBMA合金的增容作用。 首先,通过共聚反应合成了呋喃功能化的PBMA(P(BMA- co-FMA)),通过官能化改性商业PS的方法合成了马来酰亚胺功能化的PS(MPS);然后通过溶液共混方法制备了P(BMA- co-FMA)/MPS合金,通过控制两种基团的DA反应时间,研究了共混物相结构的变化,探索了PS/PBMA合金相容性提高的原因。
Bruker AV 400型核磁共振波谱仪以及Bruker AVANCE Ⅲ 400宽腔固体超导核磁共振波谱仪(NMR,德国Bruker公司);TOSOH HLC 8220型凝胶渗透色谱仪(GPC,日本TOSOH公司);Perkin-Elmer DSC7型差示扫描量热仪分析(DSC,美国Perkin-Elmer公司);JEM1011型透射电子显微镜(TEM,日本JEOL公司);Leica Ultracut R型超薄切片机(德国Leica Microsystems公司);Instron 1121型材料试验机(英国Instron公司)。
PS( Mn=8.5×104,PDI=2.36)购自中国石油大庆石油化工总厂;溴化亚铜,五甲基二乙烯三胺(PMDETA)以及2-溴异丁酸乙酯(EBIB)购自Sigma-Aldrich公司。呋喃甲醇,甲基丙烯酸甲酯(MMA),氢氧化钾,甲基丙烯酸正丁酯(BMA)等试剂购自国药集团化学试剂有限公司;环己烷,甲苯,乙酸乙酯等有机溶剂购自北京化工厂。 以上试剂均为分析纯。
甲基丙烯酸呋喃甲醇酯(FMA)单体利用呋喃甲醇和甲基丙烯酸甲酯之间的酯交换反应合成,反应使用氢氧化钾作为催化剂。 具体的实验过程如下:将甲基丙烯酸甲酯(120 mL, 1.13 mol),氢氧化钾(7.2 g,0.13 mol),呋喃甲醇(55 mL,0.63 mol),环己烷(280 mL)一起放入500 mL三口圆底烧瓶中,加入聚四氟乙烯搅拌子,然后安装分水器以及球形冷凝管,分水器出水端预先装入约1/2容积的水,然后升温至110 ℃开始搅拌加热,回流反应。 酯交换反应生成的甲醇蒸发冷凝后进入分水器溶解在水中,其余馏分(甲基丙烯酸甲酯和环己烷)浮于水上方,流回圆底烧瓶中循环。回流反应进行12 h后停止加热搅拌,冷却后将瓶中液体倒入300 mL蒸馏水中,充分搅拌后静置分层,使用分液漏斗分离有机相,然后将有机相使用蒸馏水洗涤3次后,无水硫酸镁干燥,旋蒸浓缩,减压蒸馏得到44.8 g目标产物,为无色油状液体,收率61%。1H NMR(CDCl3,400 MHz), δ:7.42(dd, J=1.9,0.8 Hz,1H),6.42(d, J=3.1 Hz,1H),6.36(dd, J=3.3,1.9 Hz,1H),6.13(t, J=1.3 Hz,1H),5.57(t, J=1.6 Hz,1H),5.14(s,2H),1.95(t, J=1.3 Hz,3H)。
参照参考文献[10,11]中的方法合成 N-(氯甲基)马来酰亚胺,收率11%。1H NMR(CDCl3,400 MHz), δ:6.85(s,2H),5.34(s,2H)。
实验所用聚合物按照Scheme 1所示途径合成,所用PS为商业化样品。 MPS由付克烷基化反应合成,使用商业化的PS样品与 N-(氯甲基)马来酰亚胺在四氯化锡的催化下进行反应[12]。 采用原子转移自由基聚合(ATRP)方法制备BMA均聚物及其共聚物。 在N2气气氛下将BMA和甲苯注入装有搅拌子和回流冷凝管的两口圆底烧瓶中,然后将配制好的溴化亚铜和PMDETA的溶液加入,最后注入引发剂EBIB。 将上述圆底烧瓶放入80 ℃油浴中加热搅拌开始反应,反应48 h后。将瓶中液体用乙酸乙酯稀释后通过中性氧化铝柱子除去其中的催化剂,然后用甲醇沉淀,所得固体在40 ℃下真空干燥。
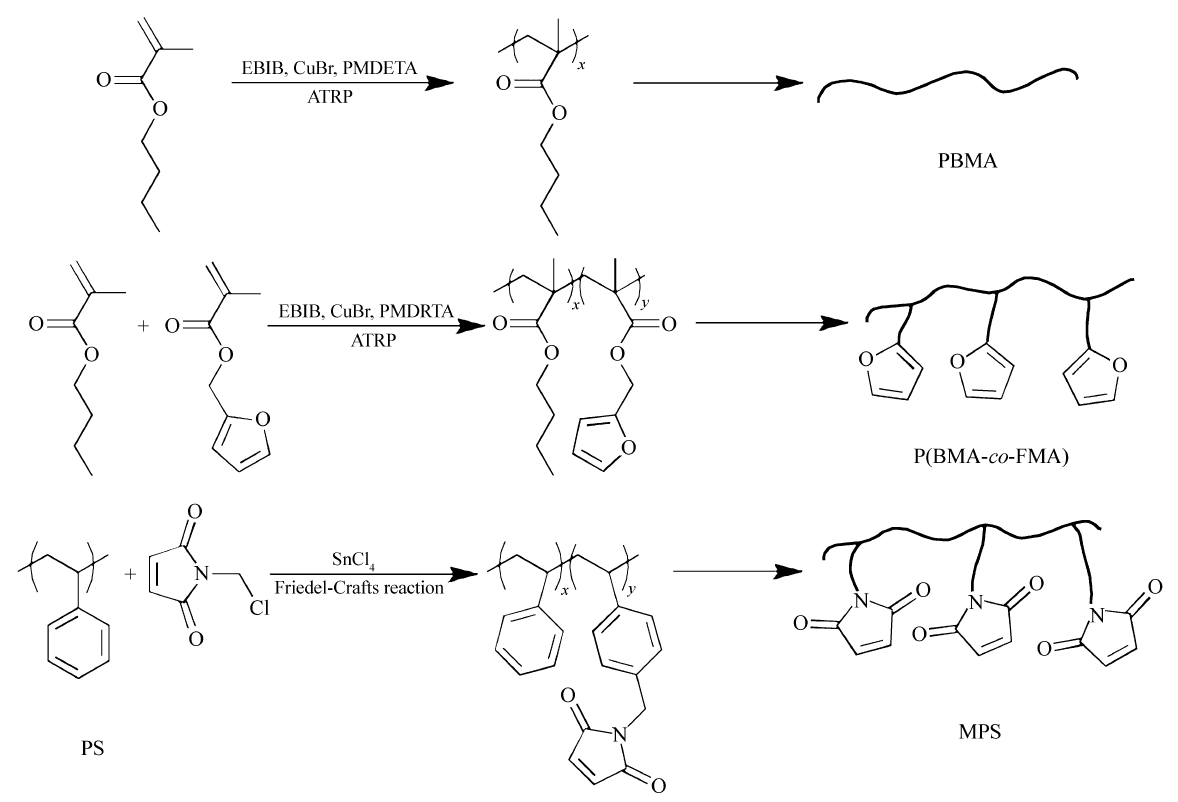 | Scheme 1 Synthesis of functionalized polymers |
{kind=link}
BMA和FMA的共聚反应其方法与BMA的均聚相同,使用溴化亚铜和PMDETA作催化剂,使用EBIB作为引发剂在甲苯溶剂中进行。
为了更直观的验证P(BMA- co-FMA)和MPS上的呋喃和马来酰亚胺基团的热可逆DA反应的发生,将这两种高分子分别与含有对应的官能团的小分子化合物进行反应以观察其DA反应的发生状况。 具体实验如下:将P(BMA- co-FMA)(20.5 mg,含有呋喃基团0.016 mmol)和 N-苄基马来酰亚胺(3.56 mg,0.019 mmol)、MPS(20.7 mg,含有马来酰亚胺基团0.013 mmol)和乙酸呋喃甲醇酯(2.94 mg,0.021 mmol)分别放入两支核磁管中,各加入氘代邻二氯苯0.5 mL然后在150 ℃油浴中加热2 h使固体充分溶解后进行核磁表征。 之后样品进行交替变温加热处理后进行核磁表征(60 ℃,48 h;150 ℃,8 h)。
功能化的MPS与P(BMA- co-FMA)之间的反应在溶液中进行。 首先,将质量比为1:1的MPS与P(BMA- co-FMA)溶解在乙酸乙酯中得到0.1 g/mL的溶液,然后使用高纯N2气吹扫该溶液10 min后在N2气保护下置于70 ℃油浴中加热搅拌。 分别反应0、9和48 h后,将反应混合物倒入甲醇中搅拌,过滤后,在室温下真空干燥72 h。
为了与MPS/P(BMA- co-FMA)合金进行对比,还制备了其余3种样品:PS/P(BMA- co-FMA)[ m(PS): m(P(BMA- co-FMA)=1:1],MPS/PBMA[ m(MPS): m(PBMA)=1:1],PS/PBMA[ m(PS): m(PBMA)=1:1],其制备条件与MPS/P(BMA- co-FMA)相同(70 ℃加热搅拌48 h)。
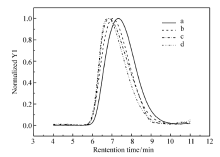
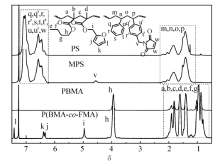
图1和图2分别为所用高分子的GPC谱图和1H NMR谱图。表1列出了聚合物样品的基本结构参数。 PS的马来酰亚胺功能化通过傅克反应(Friedel-Crafts reaction)实现。 得到的MPS样品中马来酰亚胺官能度通过1H NMR结果进行计算(图2),所得MPS的官能度为7%(摩尔分数 x%)。图1为所合成样品的GPC曲线。
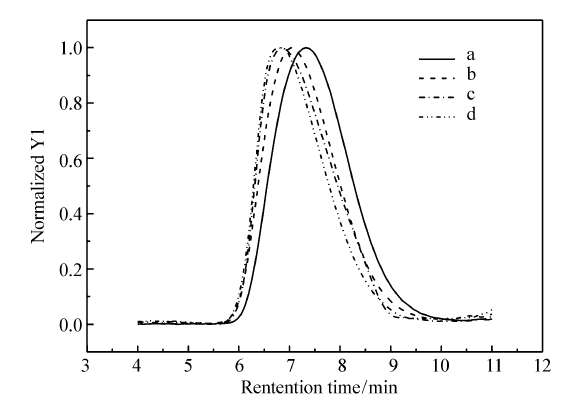 | 图1 所用高分子的GPC谱图Fig.1 GPC traces of all the polymers a.PS; b.MPS; c.PBMA; d.P(BMA- co-FMA) |
{kind=link}
 | 图2 所用高分子的核磁(氢谱)谱图Fig.2 1H NMR spectra of all the polymers in CDCl3 a.PS; b.MPS; c.PBMA; d.P(BMA- co-FMA) |
{kind=link}
| 表1 高分子的基本性质表征 Table 1 Molecular parameters of all the polymers |
所得PBMA和主链上连接有呋喃基团的P(BMA- co-FMA)通过GPC,1H NMR进行表征。通过3.94和4.95处的吸收峰(图2)的积分值对比,可以得出P(BMA- co-FMA)中的FMA摩尔分数为11.4%,与聚合反应时的投料比接近(FMA摩尔分数11%),表明在聚合反应中FMA和BMA的反应活性接近。
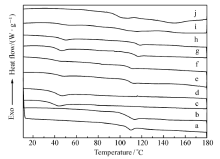
图3为所有高分子及其共混物的DSC表征结果。 由于呋喃和马来酰亚胺基团的引入,P(BMA- co-FMA)的玻璃化温度( Tg)由DSC测得为41.0 ℃,比PBMA的 Tg(38.6 ℃)略高,而MPS的 Tg为107.4 ℃,也略高于PS的 Tg(106.8 ℃)。
 | 图3 聚合物及其所形成的共混物的DSC表征Fig.3 DSC curves of all the polymers and blends a.PS; b.MPS; c.PBMA; d.P(BMA- co-FMA); e.P(BMA- co-FMA)/PS, 70 ℃, 48 h; f.PBMA/PS, 70 ℃, 48 h; g.PBMA/MPS, 70 ℃, 48 h; h.P(BMA- co-FMA)/MPS, 70 ℃, 0 h; i.P(BMA- co-FMA)/MPS, 70 ℃,9 h; j.P(BMA- co-FMA)/MPS, 70 ℃,48 h |
{kind=link}
通过乙酸乙酯溶液共混的方法制备了4种高分子合金:PS/PBMA、PS/P(BMA- co-FMA)、MPS/PBMA和MPS/P(BMA- co-FMA),组成比均为1:1(质量比)。 这4种高分子合金溶液均于N2气气氛下在70 ℃中加热48 h,发现MPS/P(BMA- co-FMA)体系在加热24 h后变为凝胶状,其余3种高分子合金未发生明显变化。 首先,采用DSC表征了共混体系的相容性。 由图3可知,PS/PBMA、PS/P(BMA- co-FMA)和MPS/PBMA高分子合金体系均有两个玻璃化温度( Tg),分别与其组成中两种聚合物的 Tg相同,表明这3种高分子合金均为不相容的。 MPS/P(BMA- co-FMA)合金与前3种高分子合金的情况不同,该合金具有一个位于98.2 ℃的 Tg,介于其两种组分MPS和P(BMA- co-FMA)的 Tg之间;同时,在140~160 ℃出现一个吸热峰。 上述结果表明,MPS与P(BMA- co-FMA)两种组分间具有相容性。 另外,其140 ℃至160 ℃的吸热峰与呋喃/马来酰亚胺发生DA反应得到的产物的分解温度范围吻合[13],表明这2种高分子之间发生了DA反应。 与其余3种高分子合金对比,证明呋喃和马来酰亚胺之间的DA反应可以有效地提高PS和PBMA之间的相容性。 当MPS和P(BMA- co-FMA)在乙酸乙酯溶液中共混不加热时,共混物可以观察到2个 Tg温度(图3),一个位于41.0 ℃,另一个位于107.4 ℃,分别与P(BMA- co-FMA)和MPS的 Tg相同,表明共混物中两种组分不相容,有相分离结构的形成,TEM表征结果验证了DSC结果(图4)。 而且,此共混物的DSC表征也未观察到其余明显变化。 当MPS和P(BMA- co-FMA)在乙酸乙酯溶液中共混并且在70 ℃下加热9 h后,该共混物也具有两个玻璃化温度,分别与其组分的玻璃化温度接近,但是略微相互靠近,同时140 ℃到160 ℃的吸热峰开始出现。
 | 图4 4种高分子合金的透射电镜表征Fig.4 TEM images of four polymer blends(1:1 mass ratio) heated at 70 ℃ for 48 h in ethyl acetate A.PBMA/PS blend; B.PBMA/MPS blend; C.P(BMA- co-FMA)/PS blend; D.P(BMA- co-FMA)/MPS blend |
{kind=link}
为了深入研究高分子合金的相结构,使用TEM观察合金样品的超薄切片方法以获得直观的相结构信息。 由图4可知,当在70 ℃的乙酸乙酯溶液中加热48 h后,PBMA/PS、PBMA/MPS和P(BMA- co-FMA)/PS均具有明显的相分离结构(图4A~4C),且相结构非常类似,而MPS/P(BMA- co-FMA)在本实验条件下未观察到相分离结构(图4D)。 TEM结果表明,前3种共混物是不相容的,而MPS/P(BMA- co-FMA)是相容的,与DSC表征结果相符合。 由图5可知,当MPS/P(BMA- co-FMA)在乙酸乙酯溶液中混合后不加热马上沉淀并且在室温下真空干燥后,有明显的相分离现象出现(图5A),此时的DSC表征观察到该共混物具有两个 Tg,且与共混物中两组分的两个玻璃化温度相同,表明此时共混物中未发生DA反应。 当MPS/P(BMA- co-FMA)在70 ℃的乙酸乙酯溶液中加热9 h后,在本实验条件(放大倍数3万倍)下未观察到明显的相分离结构,但存在微小的相分离结构(图5B),同时DSC表征观察到共混物仍具有两个 Tg,且在140~160 ℃区域出现DA反应产物的分解吸热弱峰,表明这两种高分子之间发生了一定程度的DA反应。 当加热48 h后,在本实验条件(放大倍数3万倍)下TEM未观察到相分离结构,同时DSC表征也观察到仅一个介于两组分 Tg之间的玻璃化温度,同时有更明显的DA反应产物分解吸热峰,表明该共混物的两种组分相容性得到显著提升。 通过DSC和TEM表征,证明呋喃和马来酰亚胺之间的DA反应能明显改善PS和PBMA的相容性。
 | 图5 P(BMA- co-FMA)/MPS合金(1:1质量比)在70 ℃下乙酸乙酯溶液中加热不同时间后的TEM照片Fig.5 TEM images of the P(BMA- co-FMA)/MPS blend(1:1 mass ratio) heated in ethyl acetate at 70 ℃ for 0 h(A), 9 h(B) and 48 h(C) |
{kind=link}
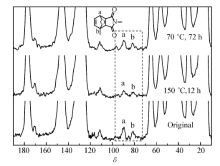
为了对P(BMA- co-FMA)/MPS中的DA/rDA反应进行直接观察,采用固体13C NMR对其进行了表征,结果如图6所示。 以体系中的DA反应形成的六元环上的两个碳(图6中a和b)的吸收峰为特征峰进行跟踪观察。
 | 图6 P(BMA- co-FMA)/MPS中的DA/rDA反应的固体13C NMR跟踪表征Fig.6 Solid13C NMR characterization of the DA/rDA reaction in the P(BMA- co-FMA)/MPS blend |
{kind=link}
当P(BMA- co-FMA)和MPS在70 ℃乙酸乙酯溶液中反应48 h后,所得产物的13C NMR谱图有明显的DA反应产物特征峰。 将该样品在150 ℃下,加热12 h后,其中DA反应产物的特征峰明显减弱,但并未消失,表明此时体系中仍然有部分DA连接处于连接状态未断开,根据文献报道[13],150 ℃加热12 h已足够使DA连接充分断裂。 但是,该体系中呋喃和马来酰亚胺连接于不同的高分子上,且原先不相容的两种高分子此时处于完全相容的状态(DSC仅观察到一个玻璃化温度,TEM观察不到相分离结构),表明当该样品在固体状态或熔体状态下进行反应时,链缠结以及交联作用导致体系中的呋喃基团与马来酰亚胺基团位置非常接近,所以逆DA(rDA)反应能够发生但是不如常见的“功能化高分子+小分子交联剂”体系以及完全由功能化小分子单体构成的体系中rDA反应的效率高。 当将该样品继续在70 ℃下加热72 h后,可以观察到体系中DA反应产物的特征峰完全恢复,因此证明该体系中的DA连接在本实验条件下具有可逆性。
为了进一步验证P(BMA- co-FMA)和MPS中的呋喃基团和马来酰亚胺基团间可逆DA反应,采用了模型反应的方法,使用含有相应官能团的小分子化合物 N-苄基马来酰亚胺和乙酸呋喃甲醇酯分别与P(BMA- co-FMA)和MPS在不同温度下进行反应,并且通过原位的1H NMR来检测DA反应和rDA反应的发生。 反应在核磁管中不同温度下进行,使用氘代邻二氯苯作为溶剂,核磁结果如图7所示。 样品装入核磁管后首先在150 ℃下加热2 h使固体充分溶解,此时在图7A和7B中的曲线a上观察不到DA反应产物的特征峰。 将样品在60 ℃下加热48 h后,在图7A和7B中的曲线b上观察到有DA反应产物的特征峰出现,表明呋喃和马来酰亚胺基团之间发生了DA反应。再在150 ℃下加热8 h后,图7A和7B中的曲线c上该特征峰消失,表明有rDA反应发生。将样品重新在60 ℃下加热48 h后,在图7A和7B中的曲线d上,该特征峰重新出现。 上述结果表明,两种功能化高分子中的呋喃和马来酰亚胺基团均能与对应的基团发生可逆的DA反应。
 | 图7 功能化高分子的模型反应1H NMR谱Fig.7 1H NMR spectra of the model reaction of the functionalized polymers A.P(BMA- co-FMA) with the N-benzyl maleimide; B.MPS with the furfuryl acetate |
{kind=link}
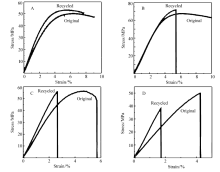
本工作中的P(BMA- co-FMA)/MPS合金使用呋喃/马来酰亚胺的DA反应进行增容。 通过P(BMA- co-FMA)和MPS分别与含有对应官能团的小分子化合物进行模型反应并通过1H NMR对其进行跟踪观察可以发现P(BMA- co-FMA)和MPS中的呋喃和马来酰亚胺基团均可进行可逆的DA反应且未观察到其它反应的发生。 通过固体13C NMR对不同温度下处理后的P(BMA- co-FMA)/MPS合金进行检测也可以观察到有可逆的DA反应发生。 因此,我们对P(BMA- co-FMA)/MPS合金的再加工性能进行了验证,并使用三点弯曲实验对其性能进行表征。 三点弯曲实验在Instron 1121型材料试验机上进行,样品尺寸为2 mm×10 mm×40 mm,支座间距30 mm,压速5 mm/min。 结果如图8所示。 研究发现,PBMA/PS合金进行再加工后其三点弯曲性能没有本质上的变化,均表现为塑性材料。 所制备的P(BMA- co-FMA)/MPS合金均可进行再加工,且随着在70 ℃乙酸乙酯溶液中反应时间的不同而表现出不同的性质。 随着2种高分子的反应时间的加长,其反应程度加大,所得高分子合金的交联程度加深,其脆性也逐渐变大。 反应0 h所得合金表现为塑性材料,发生屈服后未立即断裂,应变量到达8%时依然未断裂(实际测试中应变量高达30.0%时方发生断裂)。 反应9 h所得合金发生屈服后很快断裂,断裂时应变量为5.7%,脆性较反应0 h时增大;反应48 h所得合金完全为脆性材料,未发生屈服而直接断裂,断裂时应变量为4.3%。 将样品再次进行热压加工后,反应0 h所得合金塑性降低,屈服后很快断裂,断裂时应变量减小至5.5%,反应9 h所得合金再次加工后完全变为脆性材料,无屈服现象,应变量为2.6%时发生断裂。 反应48 h所得合金再次热压加工后应变量为1.7%时即发生断裂。
 | 图8 高分子合金在70 ℃的乙酸乙酯溶液中加热反应后的三点弯曲测试和再加工Fig.8 Three point bend tests and reprocessability of the blends heated in the solution of ethyl acetate at 70 ℃ A. PBMA/PS-heated for 48 h; B.P(BMA- co-FMA)/MPS-heated for 0 h; C.P(BMA- co-FMA)/MPS-heated for 9 h; D.P(BMA- co-FMA)/MPS-heated for 48 h |
{kind=link}
研究了呋喃/马来酰亚胺的Diels-Alder(DA)反应在P(BMA- co-FMA)/MPS共混物中增容作用。 实验发现,由于DA反应的发生,P(BMA- co-FMA)/MPS共混物能够得到有效的增容,通过控制反应时间可得到不同相结构的共混物。 呋喃功能化PBMA和马来酰亚胺功能化PS之间的模型反应结果证明了这两种高分子上的呋喃和马来酰亚胺基团能够发生可逆的DA反应,同时对P(BMA- co-FMA)/MPS合金的固体13C NMR表征也验证了高分子合金中的可逆DA反应的发生。 所得到的P(BMA- co-FMA)/MPS合金均能够进行热塑性再加工,随着DA反应时间的延长,由于交联程度的增大,所得到的高分子合金逐渐由塑性材料变为脆性材料。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|