
ZHANG Junwei, WU Hao, CHEN Zhe, et al. Recent Progress on the Construction of Chiral 3-Amino-2-oxindoles Skeleton with Isatin-Derived Ketimines as Reactants[J]. Chinese Journal of Applied Chemistry, 36(12): 1343-1360


共同通讯联系人:金瑛,教授; Tel:0432-64560318; E-mail:1364603451@qq.com; 研究方向:手性不对称催化剂合成研究及应用
手性3-氨基-2-吲哚酮骨架广泛存在于许多药物分子以及天然产物中,具有较高的药用价值。 靛红亚胺参与的不对称反应是合成手性3-氨基-2-吲哚酮衍生物的直接途径。 本文围绕着不对称aza-Henry反应、不对称环化反应及其他不对称反应3个方面,综述了靛红亚胺参与构建手性3-氨基-2-吲哚酮衍生物的研究进展并进行了展望。
Co-corresponding author:JIN Ying, professor; Tel:0432-64560318; E-mail:1364603451@qq.com
The chiral 3-amino-2-oxindole skeletons are widely present in many drug molecules and natural products, and show high medicinal value. The asymmetric reaction involving of isatin-derived ketimines provides a direct access to chiral 3-amino-2-oxindole derivatives. In this paper, the recent progress on the asymmetric aza-Henry, cyclization and other asymmetric reactions for the construction of 3-amino-2-oxindoles skeleton with isatin-derived ketimines is reviewed and the future development is prospected.
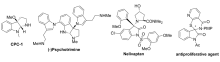
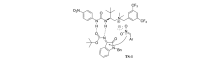
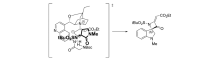
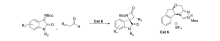


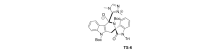


手性3-氨基-2-吲哚酮骨架广泛存在于许多药物分子以及天然产物中[1,2](图1),对于该骨架的构建最直接的手段就是以靛红亚胺作为底物,通过不对称Henry[3,4]反应、不对称Mannich[5,6,7]反应、不对称[3+2]以及[4+2]环化反应[8,9]等进行构建。 经过多年的发展,目前对于靛红亚胺参与构建手性3-氨基-2-吲哚酮骨架的方法已经发展较为成熟[10,11]。 2016年以前关于不对称催化靛红亚胺直接构建手性3-氨基-2-吲哚酮骨架的相关研究已有综述报道[12]。 因此,本文对于靛红亚胺2016年以后国内外报道的靛红亚胺参与构建手性3-氨基-2-吲哚酮骨架的不对称反应进行了综述,主要以反应类型进行分类:包括aza-Henry 反应、不对称环化反应和其它不对称反应3部分。
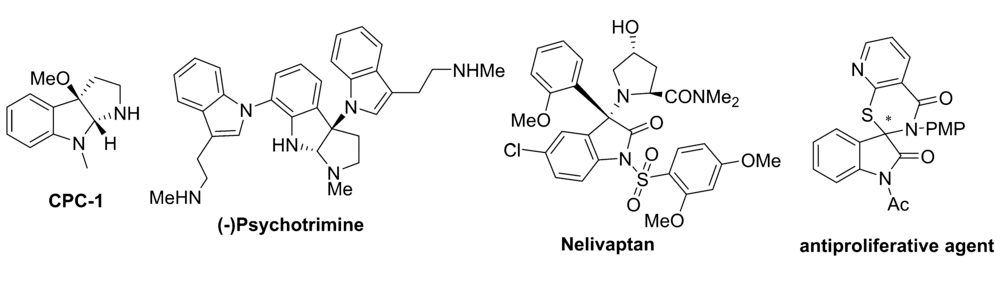 | 图1 含有3-氨基-2-吲哚酮骨架的药物分子以及天然产物化合物的实例[2]Fig.1 Examples of drug molecules and natural product compounds containing a 3-aminooxindoles skeleton[2] |
1.1.1 金鸡纳碱及其衍生物催化
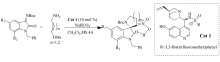
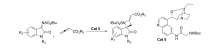
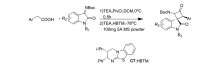
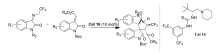
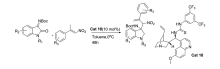
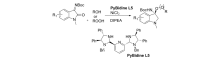
2017年,Hajra等[13]采用奎宁衍生的三官能磺酰胺催化剂Cat 1(公式中用量单位mol%指摩尔分数%)催化硝基烷-甲磺酸酯与 N-Boc靛红亚胺的aza-Henry/cyclization串联反应(图2)。 催化剂C6'-OH(羟基)对反应起着至关重要作用。 当催化剂C6'由-OH换成-OMe(甲氧基)时,产物的产率(95%→56%)和对映体过量值(90%→31% ee)显著降低。
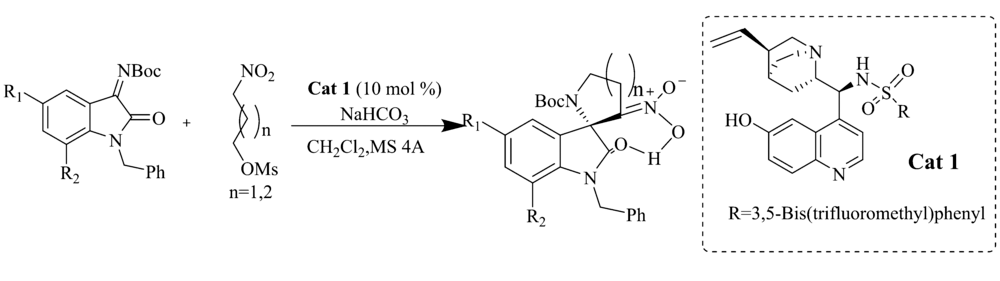 | 图2 硝基烷-甲磺酸盐与 N-Boc靛红亚胺的aza-Henry/cyclization串联反应[14]Fig.2 The aza-Henry/cyclization reaction of isatin-derived N-Boc ketimines and nitroalkane-mesylates[14] |
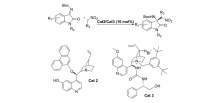
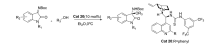
2017年,王雨卉等[14]和Liu等[15]分别报道了将催化剂Cat 2和Cat 3应用于催化 N-Boc靛红亚胺与硝基烷的不对称aza-Henry反应(图3)。 王雨卉等[14]以优秀的立体选择性(高达91% ee)构建了胺基季碳氧化吲哚骨架化合物,同时,催化剂Cat 2对靛红亚胺C1被甲基取代普适性更好,但反应时间需要3 d,而Liu等[15]以更优异的产率(96%~99%)和良好的对映选择性(高达95% ee)得到3-取代-3-氨基-2-吲哚酮骨架化合物。 除硝基乙烷(48 h)外,Cat 3催化反应时间仅需1214 h且对靛红亚胺C1被苄基取代普适性更好。
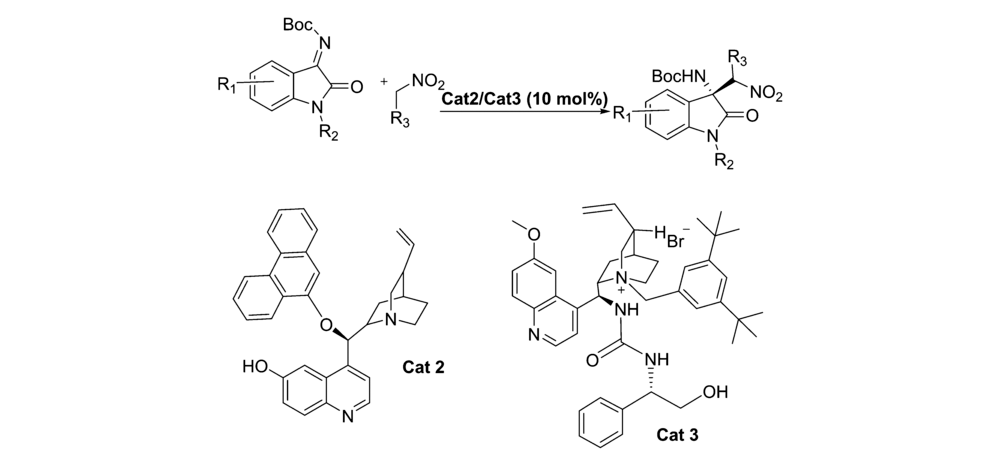 | 图3 N-Boc靛红亚胺与 α-芳基硝基甲烷的aza-Henry反应[14,15]Fig.3 The aza-Henry reaction of isatin-derived N-Boc ketimines and α-aryl nitromethanes[14,15] |
1.1.2 其它有机小分子催化
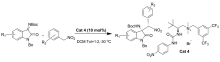
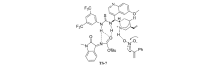
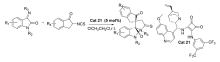
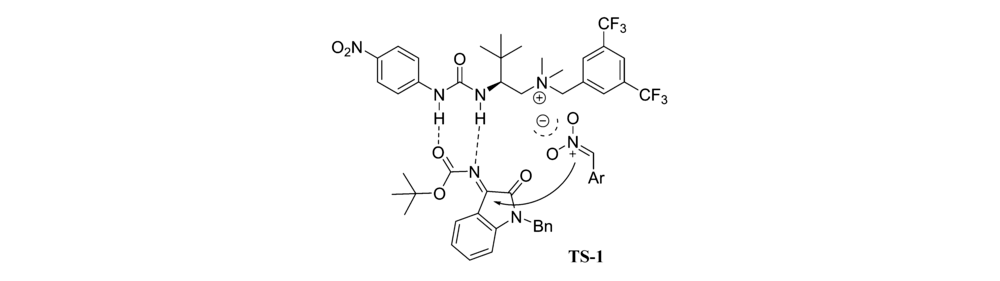
2019年,Wang等报道[16]了以 L-叔亮氨酸衍生的尿素铵盐相转移催化剂Cat 4为催化剂催化 N-Boc靛红亚胺和 α-芳基硝基甲烷的不对称aza-Henry反应(图4)。 该催化体系能以优异的产率(85%~99%)和立体选择性(79:21~97:3 dr,83%~95% ee)得到产物。 同时该反应条件较温和且可放大至克级,为此类骨架衍生物的生物活性筛选提供了可能。 推测反应机理可能是 N-Boc靛红亚胺与催化剂中NH键相互作用,亲核试剂 α-芳基硝基甲烷的硝基阴离子与催化剂中胺基通过静电作用相互配对,形成过渡态TS-1(图5),随后配对后的硝基化合物从Re面进攻过渡态TS-1,完成反应。
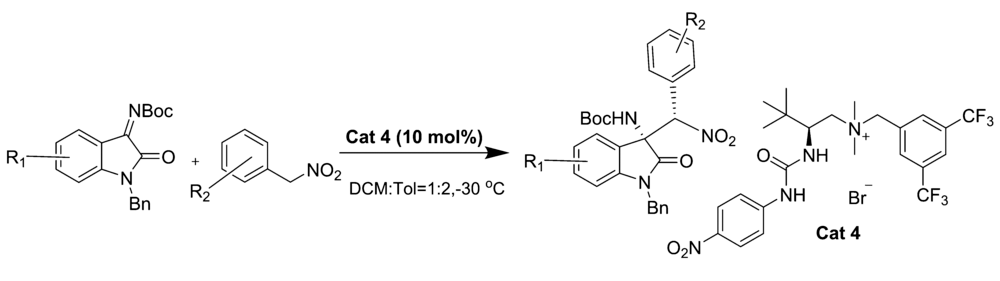 | 图4 N-Boc靛红亚胺与 α-芳基硝基甲烷的aza-Henry反应[16]Fig.4 The aza-Henry reaction of isatin-derived N-Boc ketimines and α-aryl nitromethanes[16] |
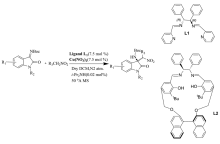
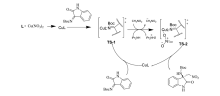
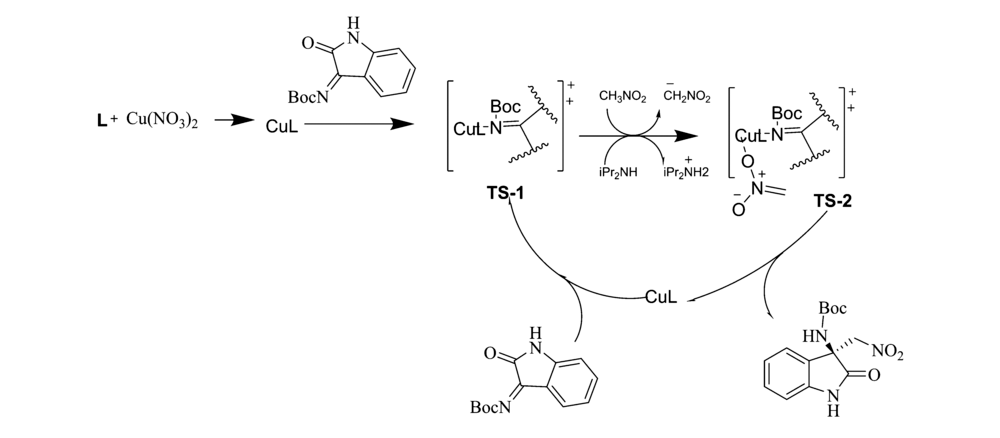
2017年,Menapara等[17]用Cu(NO3)2为催化剂,在手性含氮配体L1作用下,实现了铜(Ⅱ)催化 N-Boc靛红亚胺与硝基烷的不对称aza-Henry反应(图6)。 反应以良好的产率(65%~83%)和良好至优异的立体选择性(83%~99% ee)构建了 β-硝基胺产物。 随后,该课题组[18]又报道了将Cu(OAc)2·H2O和配体L2用于该反应中(图6)。 除7'-Cl(46% ee)外,其余反应均以较好的产率(77%~88%)和良好至优异的立体选择性(89%~99% ee)得到产物。 基于动力学数据,推断Cu(Ⅱ)催化 N-Boc靛红亚胺参与aza-Henry反应的机理为SN2亲核取代(图7)。
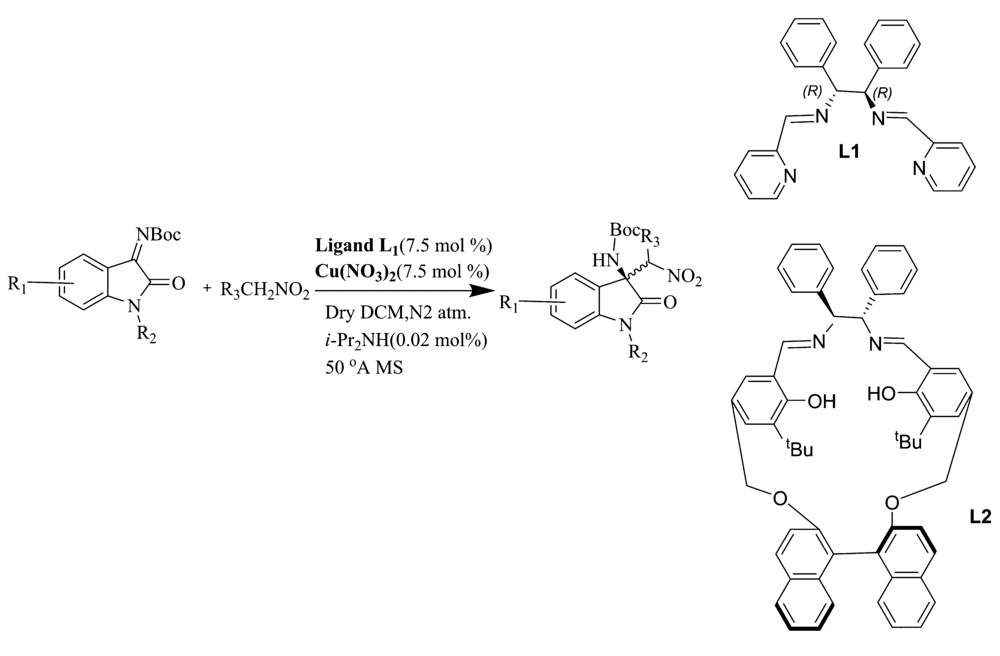 | 图6 铜(Ⅱ)催化 N-Boc靛红亚胺与硝基烷的不对称aza-Henry反应[17,18]Fig.6 Cu(Ⅱ)-catalyzed asymmetric aza-Henry reaction of isatin-derived N-Boc ketimines with nitroalkane[17,18] |
2012年,Lv等[19]首次报道应用 N-杂环卡宾(NHC)催化的靛红 N-Boc酮亚胺和烯醛的构建螺环羟吲哚- γ-内酰胺的环化反应。 此后,越来越多有机化学家参与到靛红亚胺参与的不对称环化反应催化剂的开发中。 先后报道了一系列不对称[2+2]、[3+2][20]和[4+2]等环化反应。 不同的反应当中又涉及到各种催化体系。 但目前靛红亚胺参与的[3+2]不对称环化反应报道较多。 因此,本文将重点介绍靛红亚胺参与的[3+2]不对称环化反应。
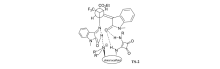
2017年,Rainoldi等[21]报道了将奎尼定衍生的生物碱 β-异哌啶催化剂Cat 5应用于的 N-叔丁基磺酰基靛红亚胺与烯酸盐的不对称[2+2]环化反应中(图8)。 以较高的产率(最高达95%)和中等立体选择性(最高66% ee)得到( S)-手性螺环吲哚基4-亚甲基氮杂环化合物。 推测反应机理为(图9):碱性奎宁环部分充当亲核试剂,与烯醇化底物产生中间体-烯丙基碳阴离子。 该中间体的活化部分向靛红亚胺添加,并分子内进攻靛红亚胺的氮,生成( S)-手性螺环吲哚基4-亚甲基氮杂环化合物和催化剂Cat 5。 此外,催化剂Cat 5中C6位存在NH和NHBoc单元可确保靛红亚胺适当活化。 依靠这种配置,可以最小化催化剂空间位阻,使靛红亚胺为进入的 γ-碳阴离子提供其Re面,得到( S)-构型中相应的螺氮杂环丁烷衍生物。 同时也解释了C6位连接NHBoc催化效果优于连接OH。
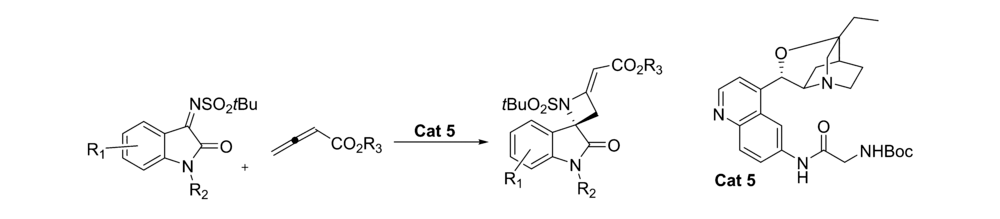 | 图8 N-叔丁基磺酰基靛红亚胺不对称环化反应[21]Fig.8 Asymmetric cyclization of N-tert-butylsulfonyl isatin-derived ketimines[21] |
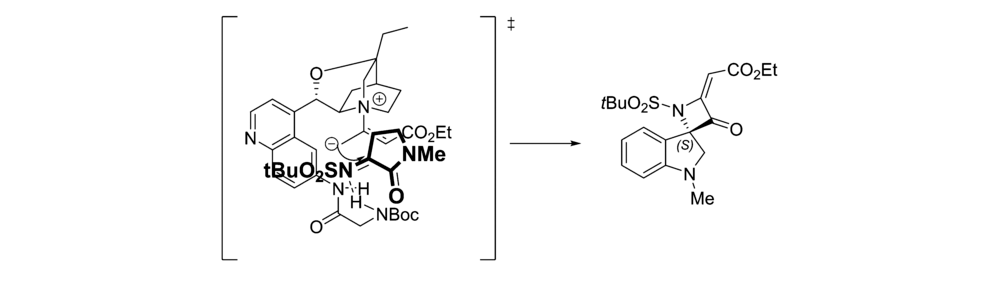 | 图9 N-叔丁基磺酰基靛红亚胺不对称环化反应反应机理[21]Fig.9 The reaction mechanism of asymmetric [2+2] cyclization of N-tert-butylsulfonyl isatin-derived ketimines[ [21] |
2017年,Xu等[22]报道了NHC催化剂Cat 6催化 N-Boc靛红亚胺与醛的不对称[2+2]环化反应(图10)。 该催化体系能以良好的产率(最高81%)和优异的立体选择性(最高为98% ee,>20:1 dr)得到带有两个邻位立体中心的(2 R,3 R)螺吲哚吲哚 β-内酰胺,并可放大至克级。
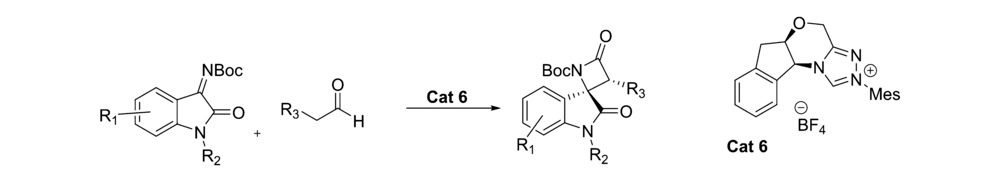 | 图10 N-Boc靛红亚胺不对称[2+2]环化反应[22]Fig.10 Asymmetric [2+2] cyclization of isatin-derived N-Boc ketimines[22] |
2018年,Guo等[23]报道了以Cu(Ⅰ)为催化剂,在手性胍-酰胺配体L3的作用下,催化靛红亚胺、苯乙炔和TsN3的叠氮炔环化/[2+2]串联反应反应(图11)。 该催化体系能以良好的产率和立体选择性得到(3 S,4 R)螺环亚酮二亚胺羟吲哚骨架化合物,且反应条件较温和,具有广谱的底物范围并可放大至克级。 结合控制实验和X射线衍射仪实验结果,推断了催化作用机制可能是:Cu(Ⅰ)与手性胍-酰胺配体L3相互作用形成簇离子对(L3·H+)2/[Cu2Cl4],该簇离子即可作为催化物质的储库,也可能直接参与催化过程。 同时手性胍-酰胺配体L3的酰胺部分与靛红亚胺之间的氢键相互作用也可能在[2+2]环化反应中起到诱导作用,导致立体构型的产生。
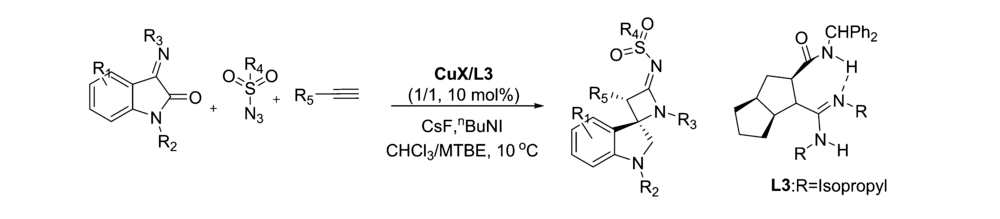 | 图11 Cu(Ⅰ)催化靛红亚胺不对称[2+2]环化反应[23]Fig.11 Cu(Ⅰ) catalytic asymmetric [2+2] cyclization of isatin-derived ketimines[23] |
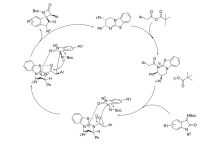
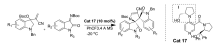
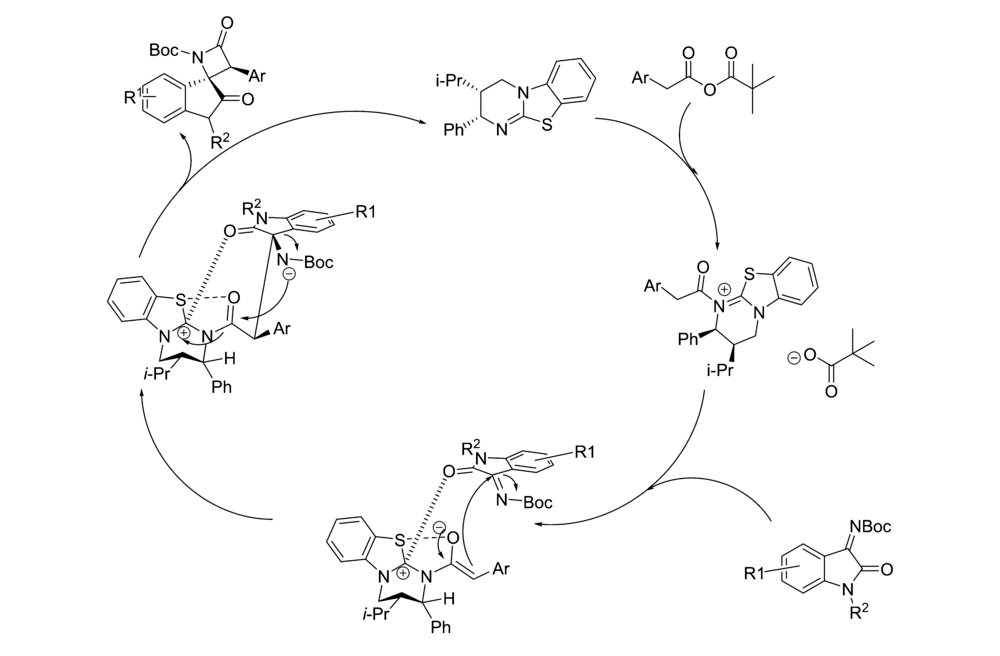
2019年,Jin等[24]将催化剂Cat 7应用于芳基乙酸和取代 N-Boc靛红亚胺不对称Mannich/lactamization反应中(图12),研究发现,该催化剂具有良好的普适性。 对于取代的芳基乙酸和取代 N-Boc靛红亚胺均能以良好的产率(75%98%)和优异的立体选择性得到产物(>99% ee)得到产物。 并认为其催化机制(图13)为:芳基乙酸与新戊酰氯反应生成混合酸酐,在与酰化HBTM形成中间体A,随后,中间体A进行去质子化,构建C1-烯醇铵结构,然后是C1-烯醇铵结构从Si面攻击 N-Boc靛红亚胺,得到中间体B,中间体B再经历分子内内酰胺化得到具有顺式螺环吲哚 β-内酰胺结构的产物。
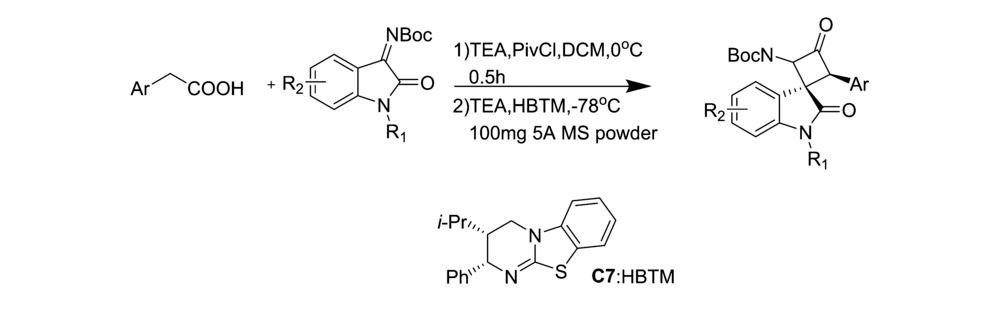 | 图12 N-Boc靛红亚胺不对称[2+2]环化反应[24]Fig.12 Asymmetric [2+2] cyclization of isatin-derived N-Boc ketimines[24] |
2.2.1 金鸡纳碱及其衍生物催化
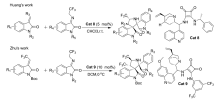
2017年,Huang等[25]首次报道了以金鸡纳碱-方酰胺类双功能催化剂Cat 8为催化剂催化亚甲基吲哚酮与靛红亚胺衍生物的不对称[3+2]环化反应中(图14),以优异的立体选择性(最高达>20:1 dr和99% ee)得到一系列3,3'-吡咯烷基-二螺吲哚化合物。 并推断靛红亚胺与催化剂Cat 8中的N—H通过双氢键活化、取向,而亚甲基吲哚酮被催化剂的叔胺活化得到过渡体TS-2(图15)。 诱导随后分子内Michael/Mannich反应产生3,3'-吡咯烷基-二螺吲哚产物。 该方法能够可以放大至克级,同时能有效地将CF3基团结合到3,3'-吡咯烷基-二螺吲哚中,为含CF3的二脱氧吲哚的新药合成奠定了理论基础。 随后,2019年,Zhu等[26]报道了将催化剂Cat 9同样应用于该类反应(图14)。 当吲哚酮骨架上衍生出吸电子取代基(EWG,如F、Cl等)或给电子取代基(EDG,如Me等),可以与含三氟甲基的靛红亚胺高效地反应,以较高产率(85%99%)和良好至优异的立体选择性(80%~>99% ee,>20:1 dr)得到结构复杂的双螺吲哚产物。 而在靛红亚胺骨架上添加Cl、Me和N-Ph等取代基,亦可以以较高的产率(90%~95%)和优异的立体选择性(>20:1 dr,97%~99% ee)得到产品。
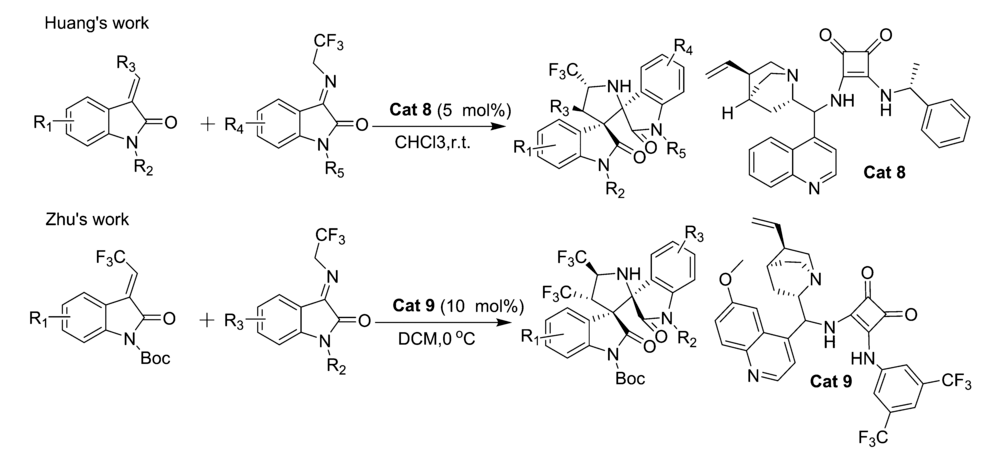 | 图14 含有CF3的靛红亚胺的不对称[3+2]环加成反应[25,26]Fig.14 The asymmetric [3+2] cycloaddition reactions of CF3-containing isatin-derived ketimines[25,26] |
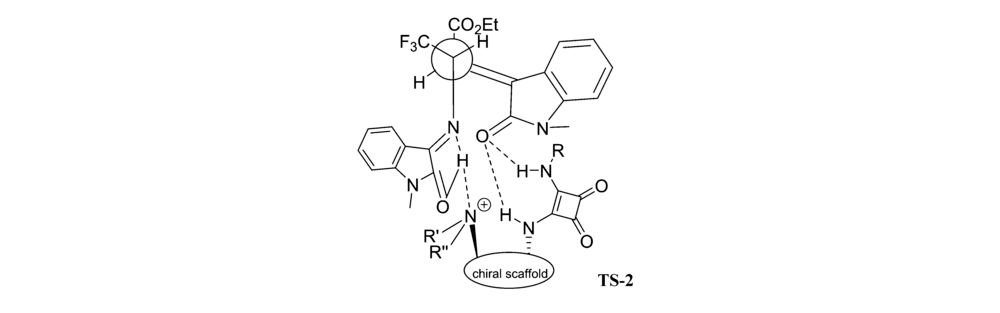 | 图15 亚甲基吲哚酮与靛红亚胺[3+2]环化反应过渡态TS-2[25]Fig.15 Transition state TS-2 of [3+2] cycloaddition of isatin-derived ketimines with methyleneindolinones[25] |
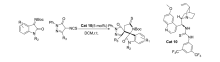
2018年,Bao等[27]报道了以金鸡纳碱-硫脲双功能类催化剂Cat 10为催化剂,二氯甲烷为溶剂,室温下催化 N-Boc靛红亚胺与4-异硫氰酸吡唑啉酮的不对称[3+2]环化反应(图16)。 该反应体系普适性好,均能以较好的产率和立体选择性得到二螺环杂环产物。 可以放大至克级。 具有(4 S,3 S')结构的产物可以经过简单的步骤转化为其他潜在的生物活性的高光学活性的手性亚砜衍生物。 为合成人类羧酸酯酶1(hCE1)抑制剂开辟了一种新方法。
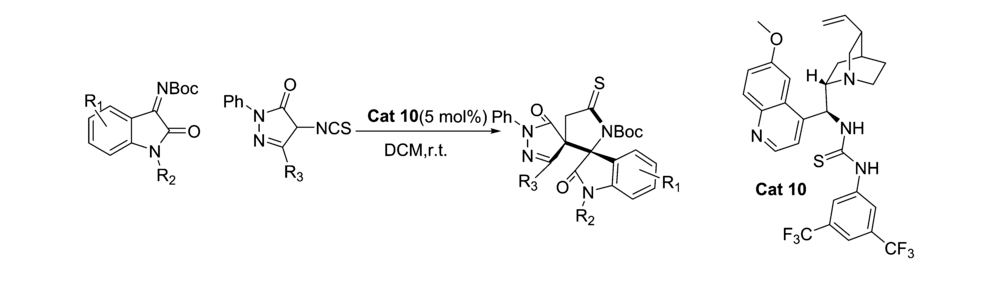 | 图16 N-Boc靛红亚胺的不对称[3+2]环化反应[27]Fig.16 The asymmetric [3+2] cycloaddition reactions of CF3-containing isatin-derived N-Boc ketimines[27] |
2018年,You等[28]报道了将Cat 11应用于催化( E) -β-三氟甲基烯酮和 N-2,2,2-三氟乙基靛红亚胺的不对称[3+2]环化反应中(图17)。 研究发现 β-三氟甲基烯酮 α'位置的芳环对反应性至关重要。 反应体系对杂芳环和萘取代的 β-三氟甲基烯酮具有很好的适应性,但不适合在 α-位带有烷基(-Bn)、烷氧基(-OEt)或酰胺基(恶唑烷-2-酮),反应可以放大至克级。 同时研究了应用催化剂Cat 12催化3-三氟乙烯吲哚酮与 N-2,2,2-三氟乙基靛红亚胺的不对称[3+2]环化反应。 反应能以优异的产率(85%97%)和立体选择性(87%95% ee,>20:1 dr)得到具有邻位双(三氟甲基)取代的3,2'-吡咯烷基螺环氧吲哚骨架的化合物。 为研究此类生物潜在的生物活性奠定了基础。
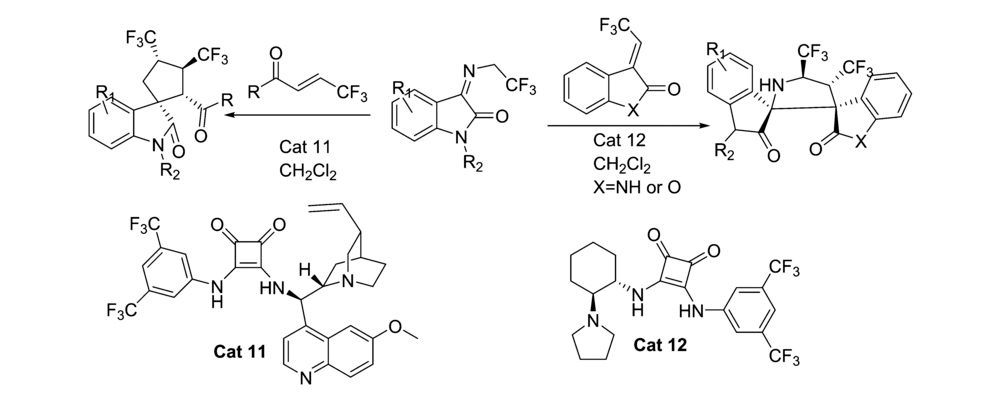 | 图17 含有CF3的靛红亚胺的不对称[3+2]环化反应[28]Fig.17 The asymmetric [3+2] cycloaddition reactions of CF3-containing isatin-derived ketimines[28] |
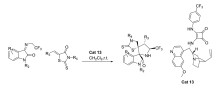
2018年,Song等[29]报道了将金鸡纳碱-方酰胺双功能催化剂Cat 13应用于罗丹宁衍生物与 N-(2,2,2-三氟乙基)靛红亚胺的不对称Domino Michael/Mannich [3+2]环加成反应中(图18)。 该催化体系能以较高的产率(最高达>99%)和优异的立体选择性(最高达>99% ee和>99:1 dr)得到具有双螺-[羟吲哚-吡咯烷-罗丹宁]骨架的产物。 该工作为具有潜在医学价值的手性杂环化合物研究提供一个新思路。
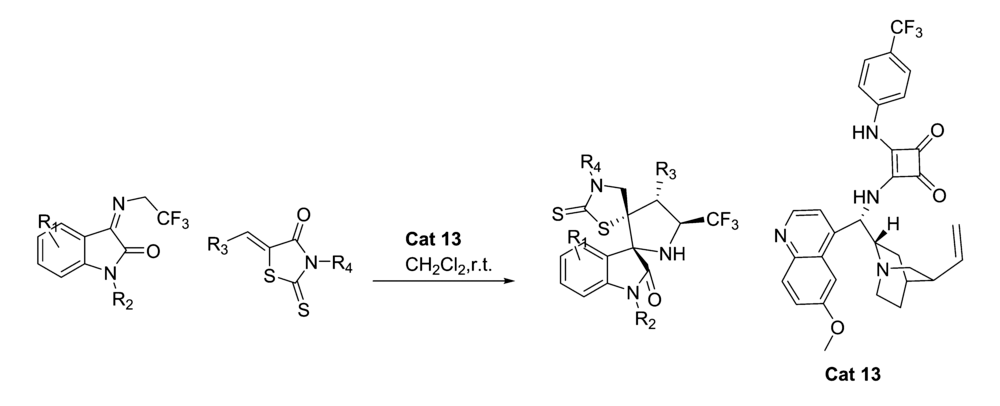 | 图18 含有CF3的靛红亚胺的不对称[3+2]环加成反应[29]Fig.18 The asymmetric [3+2] cycloaddition reactions of CF3-containing isatin-derived ketimines[29] |
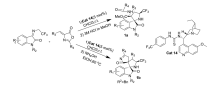
2019年,Lin等[30]报道了将金鸡纳碱衍生的硫脲类双功能催化剂Cat 14应用于 N-2,2,2-三氟乙基靛红亚胺和亚芳基吖内酯的不对称Domino Michael/Mannich [3+2]环加成反应中(图19)。 该催化体系能以60%~99%的产率和良好的立体选择性(>20:1 dr,最高达>99% ee)构建了一种含CF3的3,2'-吡咯烷基螺恶吲哚骨架结构的衍生物1a。 同时还发现,当实验条件由室温下含有3 mol/L HCl的甲醇溶液处理反应混合物改为60 ℃下用含有NH4OH的乙醇溶液处理,产物将由不稳定的3,2'-吡咯烷基螺恶吲哚骨架结构变成较稳定的3,2'-吡咯烷基二螺吲哚骨架结构化合物1b。 并且反应能够优异的对映体纯度(92%~>99% ee)得到单一非对映异构体。
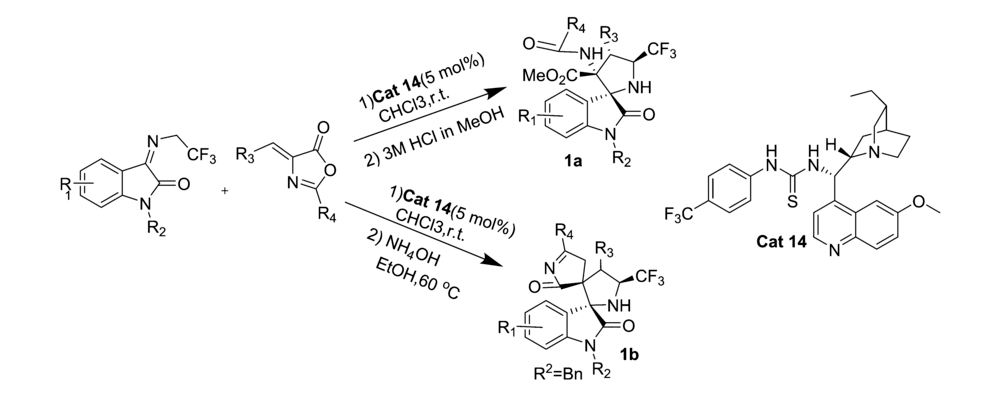 | 图19 含有CF3的靛红亚胺的不对称Domino Michael/Mannich [3+2]环加成反应[30]Fig.19 The asymmetric Domino Michael/Mannich [3+2] cycloaddition reactions of CF3-containing isatin-derived ketimines[30] |
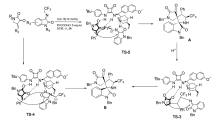
2019年,Zhao等[31]开发了一种新的非对映不对称催化催化剂Cat 15。 通过控制添加物化学计量和反应时间,可以从相同的起始原料中选择性地获得立体异构体的全部成分。 以2,3-二氧代吡咯烷和 N-2,2,2-三氟乙基靛红亚胺为模板反应,二氯甲烷为溶剂,苯甲酸为添加物。 通过控制苯甲酸化学计量和反应时间,反应均能以较高的的产率和立体选择性得到(3 S,3 'R,4 'S,5 'S)和(3 R,3 'R,4 'S,5 'S)含CF3的[双吲哚-双吡咯烷]骨架化合物(图20)。 反应体系普适性好,并能放大至克级。 根据实验数据(图21),提出了3种合理的过渡态TS-3、TS-4和TS-5,并推测反应机制为催化剂同时活化2,3-二氧代吡咯烷和 N-2,2,2-三氟乙基靛红亚胺,并诱导活化后 N-2,2,2-三氟乙基靛红亚胺从Si面进攻2,3-二氧代吡咯烷得到Michael加成产物。 随后 N-2,2,2-三氟乙基靛红亚胺的 α-碳中心攻击甲亚胺叶立德的Re面,并发生环化反应得到3 R,3 'R,4 'S,5 'S构型产物A,类似地,底物亦可通过过渡态TS-4产生另一种非对映异构体B。 或者,在反应时间内,在酸性条件下(PhCOOH),A的C1—C2键下断裂,然后经过分子内Mannich反应,经由过渡态TS-3产生B。
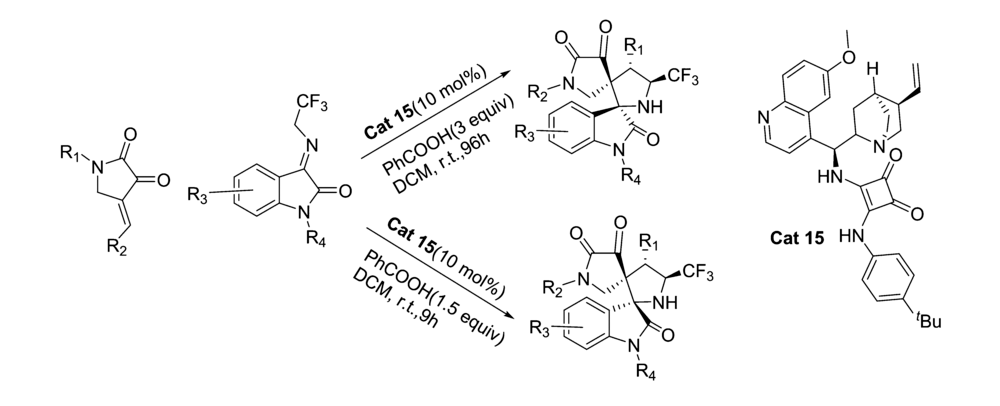 | 图20 含有CF3的靛红亚胺的不对称 [3+2]环化反应[31]Fig.20 The asymmetric [3+2] cycloaddition reactions of CF3-containing isatin-derived ketimines[31] |
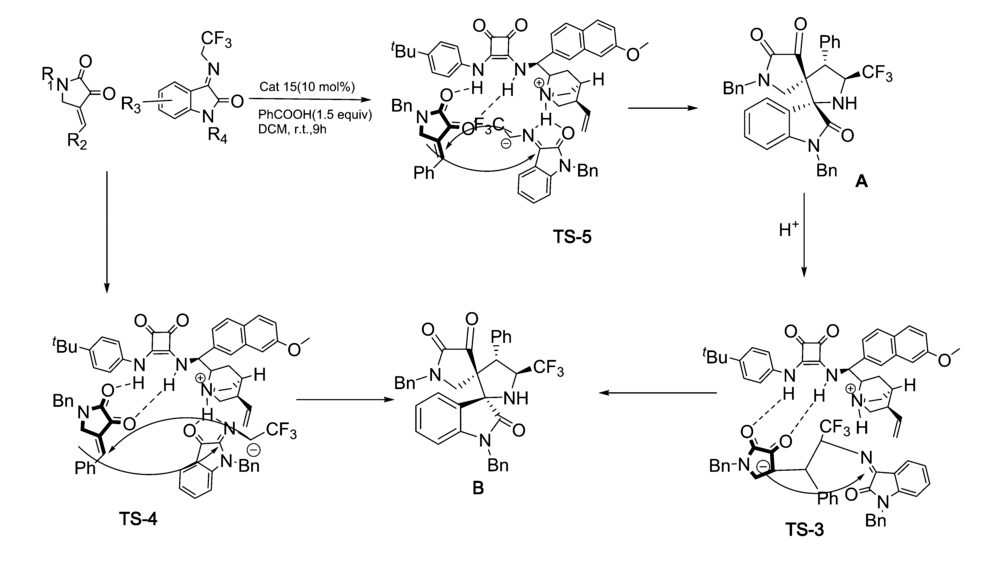 | 图21 2,3-二氧代吡咯烷和 N-2,2,2-三氟乙基靛红亚胺不对称[3+2]环化反应催化机理[31]2.2.2 其它有机小分子催化Fig.21 The mechanism of [3+2] cycloaddition reactions 2,3-dioxopyrrolidines with N-2,2,2-trifluoroethylisatin isatin-derived ketamine[31] |
2017年,Zhi等[32]报道了将硫脲类催化剂Cat 16应用于三氟乙基靛红亚胺和靛红烯酸酯的不对称Domino Michael/Mannich [3+2]环化反应中(图22)。 以优异的高光学活性合成了具有4个连续立体中心的三氟甲基化3,3'-吡咯-哌啶-二螺吲哚衍生物。 该方法为此类新药的工业化合成奠定了理论基础。
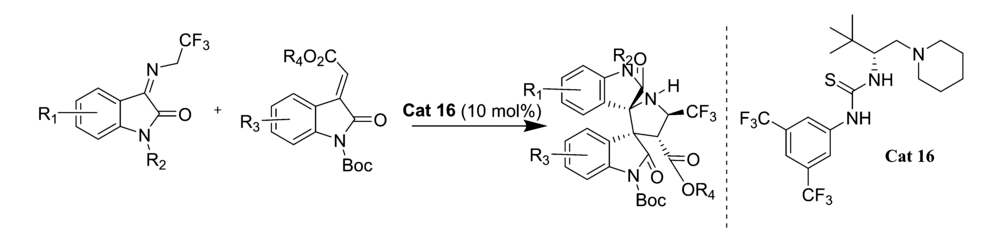 | 图22 含有CF3的靛红亚胺的不对称Domino Michael/Mannich [3+2]不对称环加成反应[32]Fig.22 The asymmetric Domino Michael/Mannich [3+2] cycloaddition reactions of CF3-containing isatin-derived ketimines[32] |
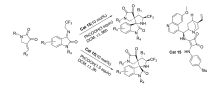
2017年,He等[33]报道了应用4-甲基氨基吡啶类物质Cat 17催化 N-Boc靛红亚胺与Morita-Baylis-Hillman盐的不对称[3+2]环化反应(图23)。 该方法能以良好的产率和中等至优异的立体选择性产生高度复杂的3,3'-吡咯烷基-二螺吲哚化合物。 催化剂中I部分的-OH对催化效果起着至关重要作用。
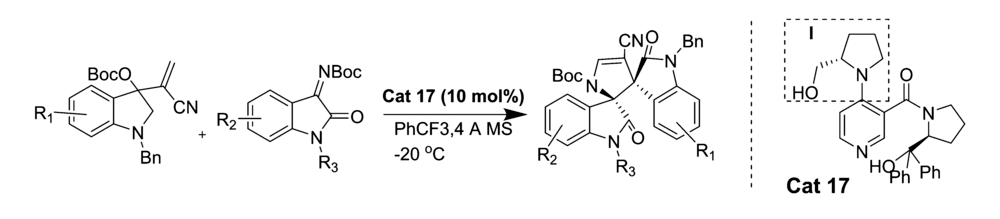 | 图23 N-Boc靛红亚胺的[3+2]不对称环加成反应[33]Fig.23 The asymmetric [3+2] cycloaddition reactions of of isatin-derived N-Boc ketimines[33] |
2019年,Wu等[34]报道了将三苯基膦应用于催化 N-2,2,2-三氟乙基靛红亚胺和 γ-取代的烯醇化合物合成螺[二氢吲哚-3,2'-吡咯]支架化合物的不对称[3+2]环化反应中(图24)。 该方法底物普适性好,可以专门用于合成含有—CF3和五元环状支架的螺环羟吲哚骨架衍生物。
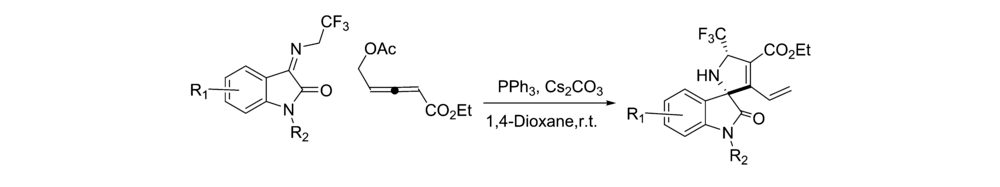 | 图24 N-Boc靛红亚胺的[3+2]不对称环加成反应[34]Fig.24 The asymmetric [3+2] cycloaddition reactions of of isatin-derived N-Boc ketimines[34] |
2.2.3 有机金属催化
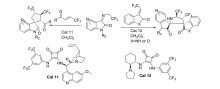
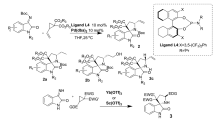
2019年,Huang等[35]报道了用Pd(dba)2作为催化剂,在手性膦配体L4作用下,实现了 N-Boc靛红亚胺与乙烯基环丙烷(VCP)的不对称[3+2]环加成反应(图25),以优异的产率和立体选择性合成了一系列螺[吡咯烷-3,2'-羟吲哚]类化合物2。 同时反应可以放大至克级。 产物2使用Pd/C作为催化剂的情况下很容易被氢化成产物2a,而在9-硼双环[3.3.1]壬烷(9-BBN)进行硼氢化并在碱性条件下用H2O2氧化可以得到伯醇类化合物2b,产品的立体选择性保持不变。 当加入在二氯甲烷中处理过的三氟乙酸时,可以容易地除去产物2中的叔丁氧基羰基,得到化合物2c,产品的对映选择性仍保持不变。 同年,Akaev等[36]开发了一种新的合成化合物3的方法(图25)。 以Yb(TOf)3或者Sc(TOf)3为催化剂,在0.1 mol/L二氯甲烷溶液中反应,得到高光学活性的螺[吡咯烷-3,2'-羟吲哚]类化合物3。 该方法对于常见的吸电子集团(EWG,如酮-、氰-、硝基-和2-吡啶基-等)和给电子集团(EDG,如Ph—、4-FC6H4、4-MeC6H4等)均能以优异的立体选择性得到具有反式C3-C5'螺[羟吲哚-3,2'-吡咯烷]化合物。
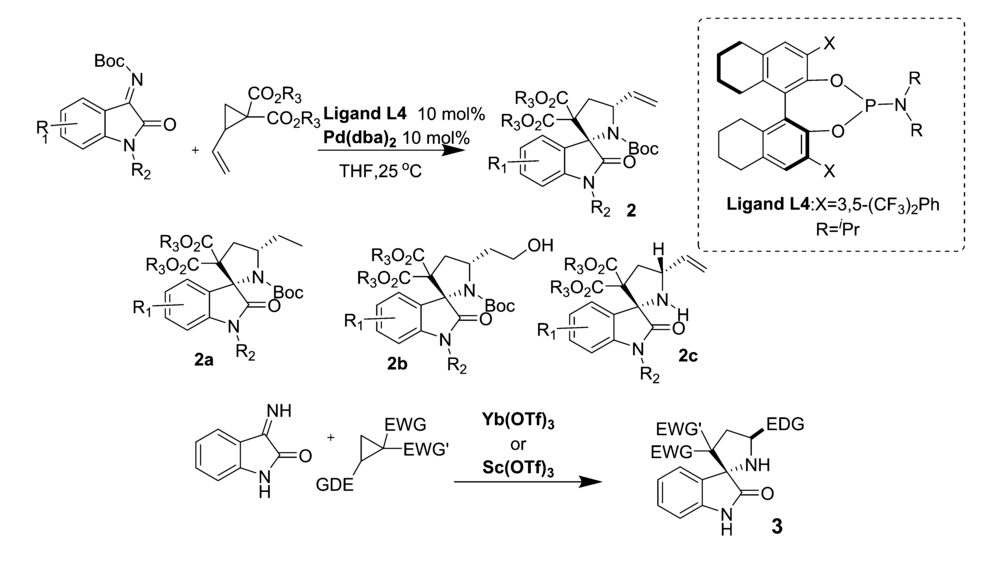 | 图25 金属催化 N-Boc靛红亚胺不对称[3+2]环化反应[35,36]Fig.25 Metal-catalyzed asymmetric [3+2] cycloaddition reaction of isatin-derived N-Boc ketimines[35,36] |
并且推断其催化机理为:类似于SN2亲核取代。 靛红亚胺首先攻击环丙烷分子的立体特异性,然后所形成高非对映选择性中间体进一步形成C—C键,获得具有高光学活性形式螺[吡咯烷-3,2'-羟吲哚]类化合物3。
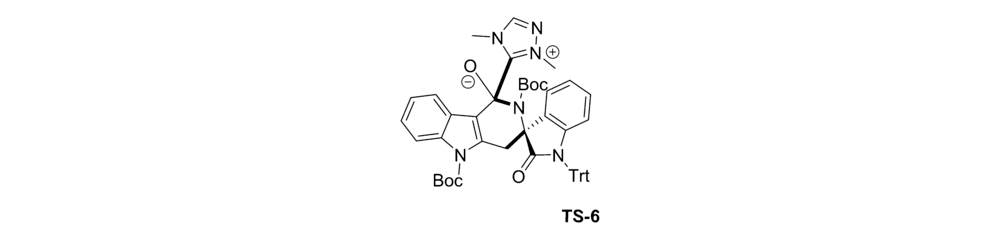
2017年,Xu等[37]报道了应用氮杂环卡宾(NHC)催化剂Cat 6催化 N-Boc-2-甲基-吲哚-3-羧酸酯原位生成杂环邻-喹啉二甲烷(oQDM)后,这些杂环oQDM与靛红亚胺发生不对称[4+2]环化反应,来构建具有高度光学活性的季铵立体中心的杂芳烃稠合的 δ-内酰胺(图26)。 该催化体系实现了C( sp3)-H直接官能化。 推测催化机制(图27)为 N-Boc-2-甲基-吲哚-3-羧酸酯与NHC结合酰基偶氮中间体,随后吲哚苄基C( sp3)去质子化,得到吲哚-2,3-喹啉二甲烷中间体,随后,该中间体与靛红亚胺进行Mannich型加成,并内酰胺化以提供过渡态TS-6。 TS-6消除NHC催化剂后得到产品。
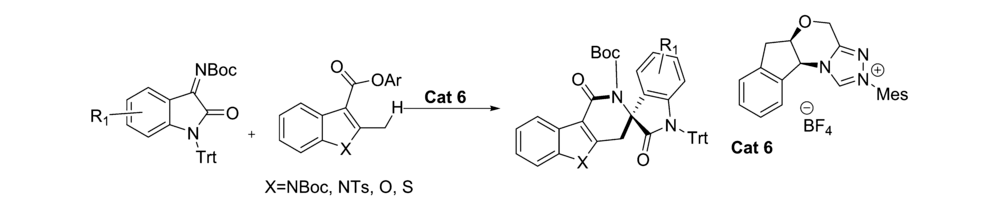 | 图26 NHC催化 N-Boc靛红亚胺不对称[4+2]环化反应[37]Fig.26 Catalytic asymmetric [4+2] cycloaddition reaction of isatin-derived N-Boc ketimines by NHC[37] |
近些年亚胺参与的不对称Aza-Friedel-Crafts反应[38,39,40]、不对称Michael[41,42]反应、不对称Morita-Baylis-Hillman 反应(不对称MBH反应),与醇的不对称加成等反应也有部分报道。 但针对靛红亚胺构建手性3-氨基-2-吲哚酮骨架的不对称Aza-MBH反应和醇的不对称亲核加成反应报道较少。 将是以后的研究方向之一。 故本文针对靛红亚胺的参与的其它不对称反应将从这两个方面介绍。
不对称MBH反应是一种生构建烯烃 α-位加成产物的有效方法。 2013年,Hu等[43]首次报道应用有机小分子催化的靛红 N-Boc酮亚胺和丁烯酮(MVK)的不对称MBH反应。 自此,有机催化靛红亚胺的不对称MBH反应也有少量报道。
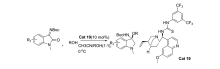
2017年,Choudhary等[44]报道了将Cat 18应用于催化 N-Boc靛红亚胺与硝基烯烃的不对称MBH反应(图28)。 反应体系能以良好的产率和优异的立体选择性得到 β-硝基- γ-烯胺类化合物。 实验组推断反应机理为催化剂与反应物之间形成的氢键相互作用,活化并化取向,从而产生立体选择性,并提出了过渡态TS-7(图29)。
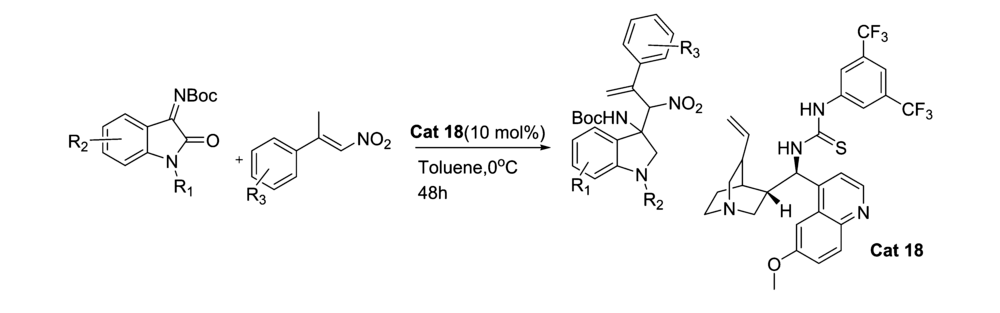
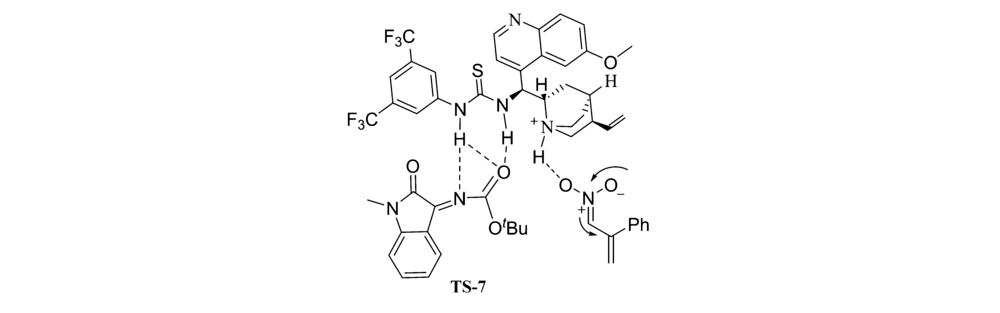
靛红亚胺与醇的不对称亲核加成反应是合成N、O缩醛胺的有效方法之一,但目前针对该方面的研究相对比较匮乏。 因此针对此研究还有很大的拓展空间。
2013年,Li等[45]首次报道了醇对 N-Boc靛红亚胺的对映选择性加成(图30)。 虽然产物的 ee值最高仅为77%,但开启了醇对靛红亚胺不对称亲和加成反应研究的序幕且该催化体系对 N-Boc靛红亚胺C1位被甲基取代的普适性良好。
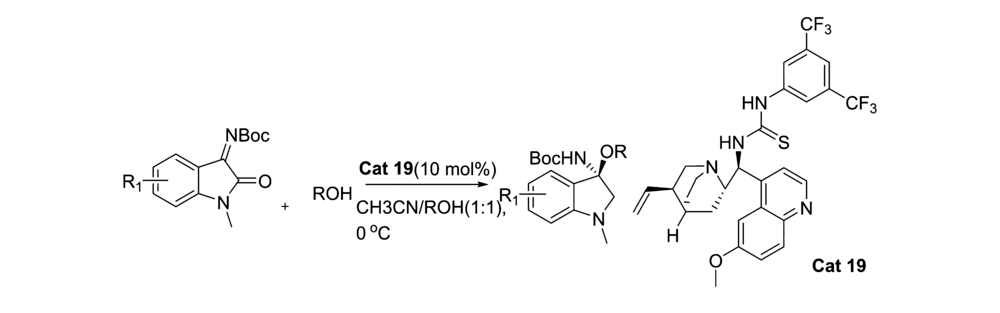 | 图30 N-Boc靛红亚胺与醇的不对称加成反应[45]Fig.30 Asymmetric addition reaction of isatin-derived N-Boc ketimines with alcohol[45] |
2015年,Aria等[46]报道了将( S, S)-二苯基二胺衍生的双咪唑烷-吡啶作为手性配体L5,不对称催化靛红亚胺与醇或过氧化物的不对称亲核加成反应(图31)。 以高达99%产率和94% ee得到C3为取代的( R)氧代吲哚。 该催化体系对 N-Boc靛红亚胺1位被甲基取代具有较好的普适性。
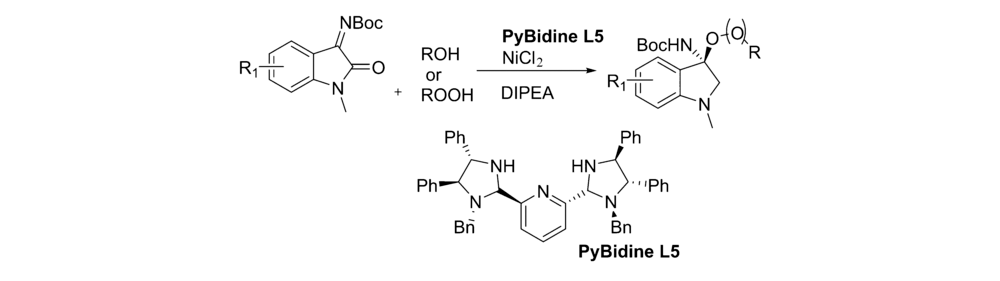 | 图31 有机金属催化 N-Boc靛红亚胺与醇的不对称反应[46]Fig.31 Asymmetric reaction of isatin-derived N-Boc ketimines with alcohol catalyzed by organometallics[46] |
2015年,刘静等[47]报道了将催化剂Cat 20应用于催化靛红亚胺与醇的不对称亲核加成反应中(图32),以较好的产率和立体选择性得到一系列3,3'-双取代氧化吲哚衍生物。 该催化方法普适性好,醇的立体结构和 N-Boc靛红亚胺苯环上的取代基对产物的立体选择性具有显著影响。
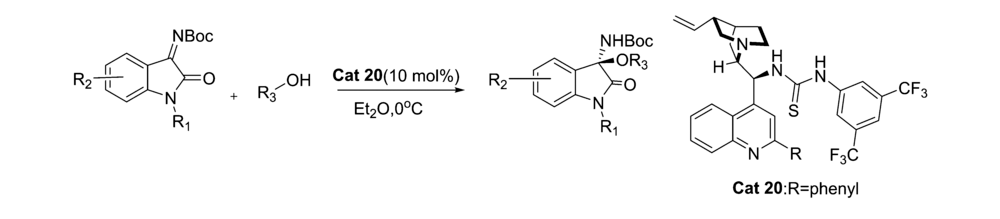 | 图32 N-Boc靛红亚胺与醇的不对称加成反应[47]Fig.32 Asymmetric addition reaction of isatin-derived N-Boc ketimines with alcohol[47] |
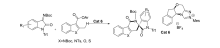
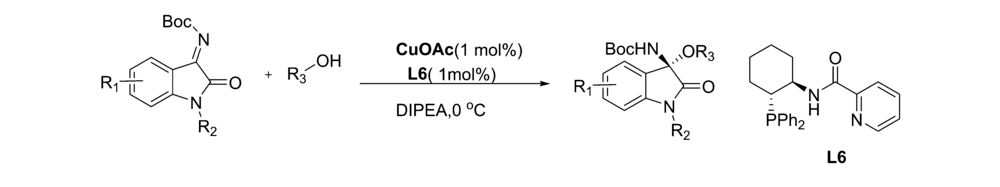
2019年,Lu等[48]报道了以Cu(OAc)2为催化剂,手性环己烷基N,P类化合物L6为配体, N, N-二异丙基乙胺(DIPEA)为溶剂,在0 ℃下催化 N-Boc靛红亚胺与醇的不对称亲和加成反应(图33)。 该方法普适性好,均能以优异的收率(93%99%)和良好至优异的对映选择性(最高达93% ee)得到手性靛红N、O缩醛胺。
2018年,Zhu等[49]报道了以CuBF4和平面手性二茂铁P,N-配体L7为复合催化剂,催化靛红亚胺与二苯亚甲基甘氨酸酯的不对称Mannich反应(图34)。 该方法普适性好,均能以优异的收率(最高为99%)和良好至优异的立体选择性(最高达>99% ee,98:2 dr)直接获得带有邻位季-叔碳立体中心的手性3-取代3-氨基-2-吲哚酮,且方法可以放大至克级。 该工作为规模化生产 α, β-二氨基酸酯提供了实验基础。
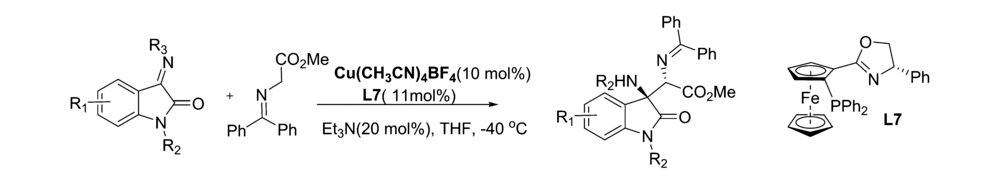 | 图34 Cu(Ⅰ)催化的靛红亚胺的不对称Mannich反应[49]Fig.34 Copper(Ⅰ)-catalyzed asymmetric Mannich reaction of isatin-derived ketimines[49] |
2018年,Zhao等[50]报道了以催化剂Cat 21不对称催化2-异硫氰基-1-茚满酮与靛红亚胺的不对Mannich/Cyclization反应中(图35)。 该催化体系能以良好的产率(最高达95%)和优异的立体选择性(最高达>25:1 dr,>99% ee)构建带有两个相邻螺-季铵立体中心的双环杂茚满-硫代咪唑烷-羟吲哚类化合物。 该催化体系反应较温和且普适性好并可以放大至克级。
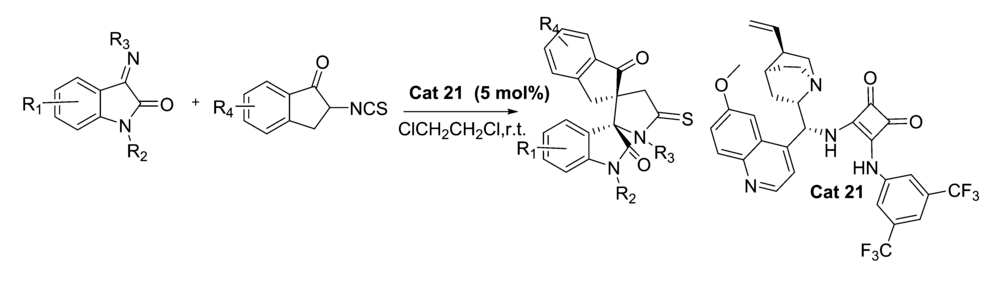
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
手性有机小分子催化下靛红亚胺的不对称反应在近年来引起了许多化学家的关注,并取得了很大进展。 主要表现在以下两个方面:1)越来越多的反应类型被发现,如不对称Aza-Henry反应、不对称环化反应、不对称Mannich反应、不对称Aza-Friedel-Crafts反应、不对称Aza-MBH反应、不对称Michael反应和醇与靛红亚胺的加成反应等,其中不对称Mannich反应报道较多。 相反,Aza-MBH反应(仅5篇报道)和醇与靛红亚胺的加成反应(仅6篇)相对较少,具有较大的研究空间。 2)催化剂的开发达到空前,包括有机小分子催化剂和有机金属催化中配体的研究。 有机催化相比于无机催化具有操作简便、产率高、立体选择性好的优点。 然而,该反应目前依然存在着很大的局限性,如普适性不高、手性催化剂缺乏等。 因此,如何开发合适的手性催化剂是研究靛红亚胺不对称反应的一个具有挑战性任务。 另外,催化剂能否与底物通过氢键相互作用对于催化反应的发生起着至关重要的作用,但机理还不够完善。 因此,研究催化剂与底物之间如何通过氢键相互作用对于催化剂的开发具有重要意义。 同时部分催化体系存在着催化剂用量多、手性金属配体过于单一、催化体系过于复杂等问题。 根据以上分析设计一些高性能、催化效果优异的手性不对称催化剂,简化催化体系必定有助于进一步完善靛红亚胺参与的不对称反应。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|