
SUN Jinyu, WANG Guilin, SHI Yufang, et al. Synthesis, Theoretical Investigation and Ultrafast Third-Order Nonlinear Optical Response of 2-(Pyren-1-yl)-1,8-naphthyridine[J]. Chinese Journal of Applied Chemistry, 36(10): 1172-1178


合成了一个供体-受体(D-A)型含芘萘啶衍生物2-(芘-1-基)-1,8-萘啶(PN)。 通过核磁共振波谱(NMR)、傅里叶变换红外光谱(FTIR)、液-质联用仪(LC-MS)表征了其结构。 通过电子光谱和Z-扫描技术方法分别研究了化合物PN的线性光学性质和三阶非线性光学吸收,采用综合热分析方法测定了它的热稳定性。 结果表明,在532 nm、180 fs条件下,PN的三阶非线性吸收行为为反饱和吸收,其吸收系数为 β=9.0×10-14 m/W,显示出超快三阶非线性光学响应。 运用密度泛函理论方法计算了分子轨道能量、极化率和超极化率,结果表明电子转移能够在分子内部进行。 2-(芘-1-基)-1,8-萘啶的紫外光谱在450 nm以上无吸收,在非线性光学吸收、激光防护、吸收型光开关或双稳器件等方面可作备选材料。
A new donor-acceptor(D-A) pyrene-containing naphthalene derivative 2-(pyrene-1-yl)-1,8-naphthyl was synthesized and characterized by nuclear magnetic resonance spectrometry(1H NMR,13C NMR), Fourier transform infrared spectrometry(FTIR) and liquid chromatography-mass spectrometry(LC-MS). The linear optical properties and third-order nonlinear optical absorption of 2-(pyrene-1-yl)-1,8-naphthyridine(PN) were studied by means of the electron spectroscopy and Z-scan technique, respectively. The thermal stability of PN was determined by thermogravimetry and differential scanning calorimetry. The experimental results show that the nonlinear absorption coefficient of PN at 532 nm and 180 fs is β=9.0×10-14 m/W, exhibiting ultrafast third-order nonlinear optical response. The molecular orbital energy, polarizability and hyperpolarizability were calculated by density functional theory, and the results show that electron transfer can take place within the molecule. There is no absorbance at more than 450 nm in ultraviolet spectrum of PN. So it is a candidate material for the next in nonlinear optical absorption, laser protection, absorption optical switch or bistable devices.

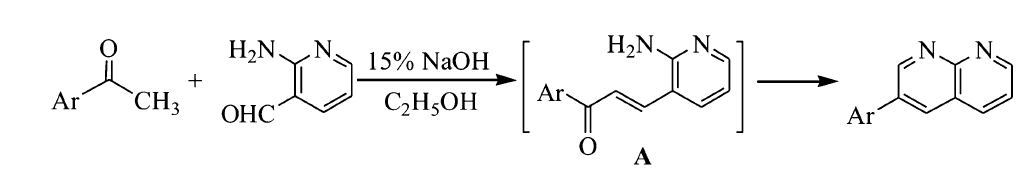
非线性光学(NLO)是科学技术的前沿领域,在激光技术[1]、光学信息处理[2]、光学数据存储技术[3]和图像处理[4]等新兴光子学应用领域中起着重要作用。 与无机材料相比,有机材料具有更高的电子极化率和分子超级化率,且更方便地调整有机分子中不同的电子供体(D)和受体(A),从而优化设计所需的NLO性能等优点[5]。 因此,有机NLO材料的设计合成及其性质研究已成为光通信[6]、光限幅[7]、光学开关[8]、数据存储[9]和动态全息[10]等领域的热门课题。 作为NLO发色团的有机共轭小分子无疑是该类材料的核心。 这些有机共轭小分子大多具有D- π-A或D-A结构,非中心对称结构的电子云有利于进行分子内电荷转移(ICT)[11]。 芘是众所周知的电子给体(D),在构建D- π-A或D-A结构的有机分子中具有自己独特的优势[12]。 但是,将两个吡啶环稠合在一起的萘啶结构单元作为电子受体(A)在有机分子设计中运用较少[13],原因在于合成该结构的困难较大。 本文在前面工作基础上,合成了一个新的D-A型芘联萘啶类化合物2-(芘-1-基)-1,8-萘啶(PN,Scheme 1),表征了其结构,并在532 nm和180 fs条件下,测定了化合物PN的三阶非线性吸收系数。
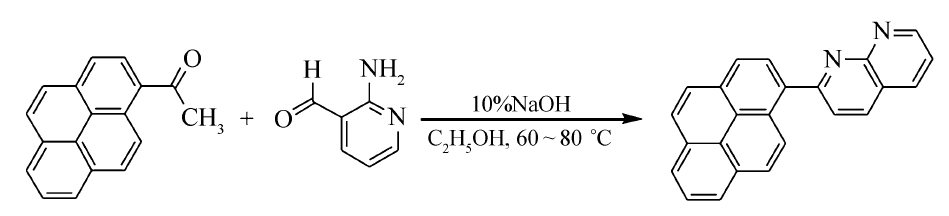 | scheme 1 Synthesis of 2-(pyren-1-yl)-1,8-naphthyridine |
UV-2550紫外可见分光光度计(UV-Vis,日本岛津公司);WRS-1B型数字熔点仪(中国上海精密科学仪器有限公司,温度未校正);F-4500型荧光光谱仪(日本日立公司);FT-IR-8400型傅里叶红外光谱仪(FTIR,日本岛津公司);Bruker Advanced III型核磁共振仪(NMR,瑞士布鲁克公司);LCMS-8030型三重四级杆液质联用仪(LC-MS,日本岛津公司),色谱条件:两通进样,流动相:A相水溶液,B相甲醇, V(A): V(B)=30:70,流速:0.4 mL/min,进样体积:5 μL;质谱条件:离子源:ESI,正离子扫描,雾化气:N2气3.0 L/min,干燥气:N2气15 L/min,碰撞气:Ar气,脱溶剂管温度:400 ℃,加热模块温度250 ℃,扫描模式:Q3Scan,驻留时间:100 ms。Nd:YAG(NT 342B)可调谐激光器(立陶宛EKSPLA公司,波长532 nm,脉宽180 fs)。 NETZSCH STA 449F3型综合热分析仪(德国Netzsch公司)。
所用试剂均为市售分析纯。
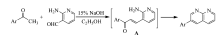
在250 mL三口圆底烧瓶中依次加入0.61 g(2.5 mmol)1-乙酰基芘(AP)、0.31 g(2.5 mmol)2-氨基吡啶-3-甲醛和90 mL无水乙醇,待固体完全溶解后加入3 mL 10%NaOH溶液,磁力搅拌下加热到60~70 ℃反应5 h,反应期间TLC跟踪反应( V(乙酸乙酯):V(石油醚)=1:3)。 反应完成后,停止搅拌,冷却,抽滤,用少量无水乙醇洗涤滤饼2次,真空干燥,得黄色粉末固体0.66 g,产率80%;mp 209.3~209.8 ℃。 IR(KBr), σ/cm-1:3037.8,1600.8,1512.1,842.8;1H NMR(600 MHz,DMSO-d), δ:9.21~9.20(t,1H, J=3.0 Hz),8.73~8.72(d,1H, J=6.0 Hz),8.65~8.63(d,1H, J=12.0 Hz),8.58~8.57(d,1H, J=6.0 Hz),8.50~8.48(d,1H, J=12.0 Hz),8.41~8.36(m,3H),8.32(s,2H),8.27~8.26(d,1H, J=6.0 Hz),8.16~8.14(t,2H, J=6.0 Hz),7.76~7.74(q,1H, J=4.0 Hz);13C NMR(150 MHz,DMSO-d), δ:162.40,155.88,154.61,138.89,138.07,135.32,131.79,131.38,130.80,128.77,128.66,128.59,127.86,127.07,126.30,125.91,125.46,125.17,125.11,124.64,124.35,123.06,121.82;LC-MS(ESI) m/z实测值:331.10;计算值C24H15N2([M+H]+):331.12。
2.1.1 FTIR光谱
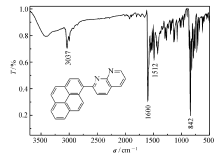
化合物PN的红外光谱如图1所示。 由图1可知,3037 cm-1为不饱和C—H的振动吸收峰,1600和1512 cm-1为苯环的特征振动吸收峰,842 cm-1为芳香环取代特征峰,图中无羰基和氨基的特征振动吸收峰,初步证明该反应产物为PN。
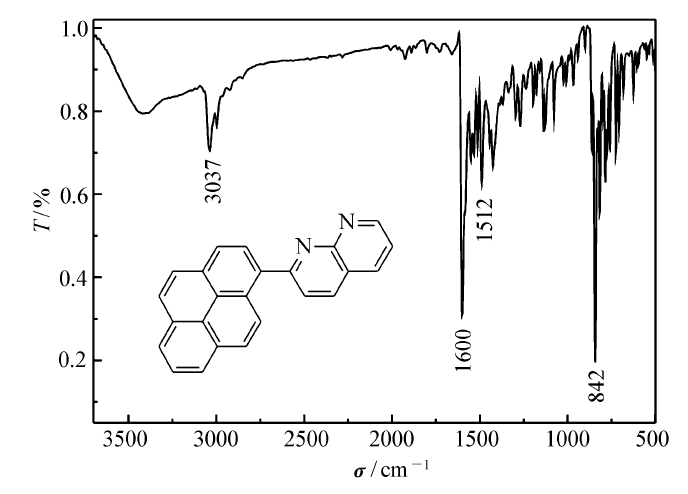 | 图1 2-(芘-1-基)-1,8-萘啶的红外光谱Fig.1 Infrared spectrum of 2-(pyren-1-yl)-1,8-naphthyridine |
2.1.2 NMR谱
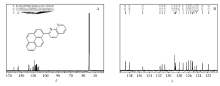
化合物PN的1H NMR谱图及其局部放大谱图如图2所示。图2A显示有14个芳香H质子种类和个数与目标化合物分子所含氢原子种类和个数相吻合。 从结构特点来说,萘啶结构由2个吡啶环稠合而成,电负性较大的氮原子使得萘啶环上氢原子的化学位移多数位于低场,而且比较密集。 9.21~9.20(t,1H, J=3.0 Hz)、8.73~8.72(d,1H, J=6.0 Hz)和7.76~7.74(q,1H, J=4.0 Hz)表示外侧吡啶环上的3个氢原子。 8.65~8.63(d,1H, J=12.0 Hz)和8.50~8.48(d,1H, J=12.0 Hz)表示内侧吡啶环上的2个氢原子。 8.58~8.57(d,1H, J=6.0 Hz)、8.41~8.36(m,3H)、8.32(s,2H)、8.27~8.26(d,1H, J=6.0 Hz)和8.16~8.14(t,2H, J=6.0 Hz)表示芘基上的9个氢原子,它们的化学位移因同属芘基而比较接近。
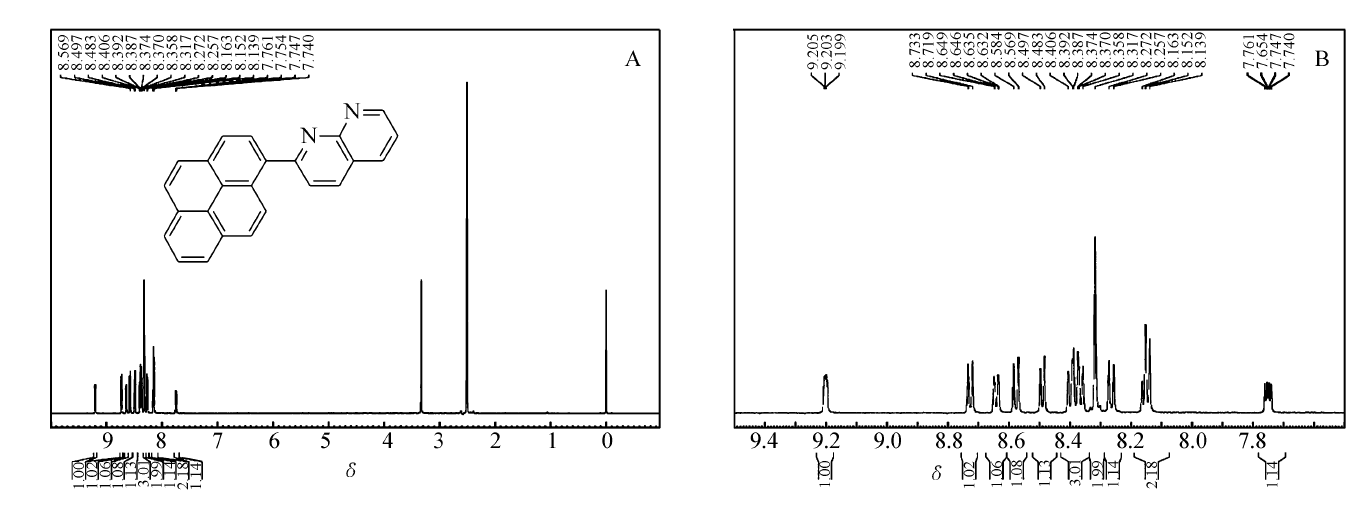 | 图2 2-(芘-1-基)-1,8-萘啶的1H NMR谱图(A)及其局部放大谱图(B)Fig.2 1H NMR(A) spectrum and partial enlarged spectrum(B) of 2-(pyren-1-yl)-1,8-naphthyridine |
化合物PN的13C NMR谱图及其局部放大谱图如图3所示。 从分子结构来看,化合物PN分子中有24种碳原子,应该出现24个碳峰。 但图3A中显示有23个碳峰,有1个碳峰隐藏。 观察13C NMR谱图的局部放大图(图3B)中各组峰的峰形,发现在128.77~128.59范围内可能隐藏1个碳峰,这是由于碳峰较多,峰位密集所导致。
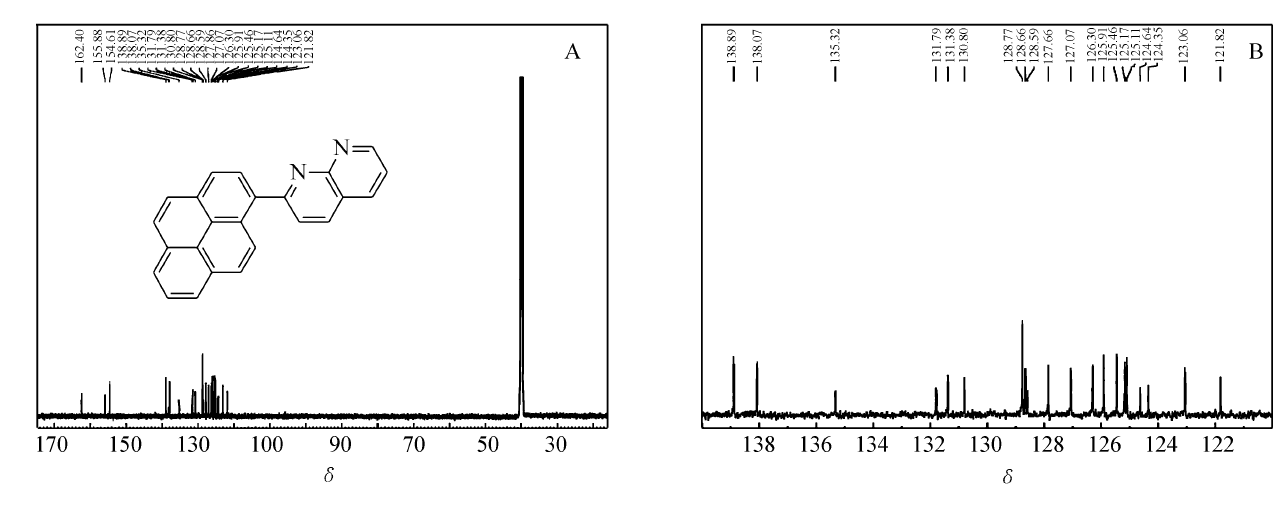 | 图3 2-(芘-1-基)-1,8-萘啶的13C NMR谱图(A)及其局部放大谱图(B)Fig.3 13C NMR(A) spectrum and partial enlarged spectrum(B) of 2-(pyren-1-yl)-1,8-naphthyridine |
2.1.3 LC-MS谱
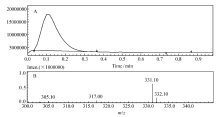
化合物PN的液相色谱和质谱如图4A和4B所示。 液相色谱(图4A)显示样品纯度很好;质谱(图4B)显示[M+H]+为331.10,与计算值(331.12)相差0.02,相对误差0.06‰,误差极小。 综合上述分析认为,合成的产物为PN,其结构正确。
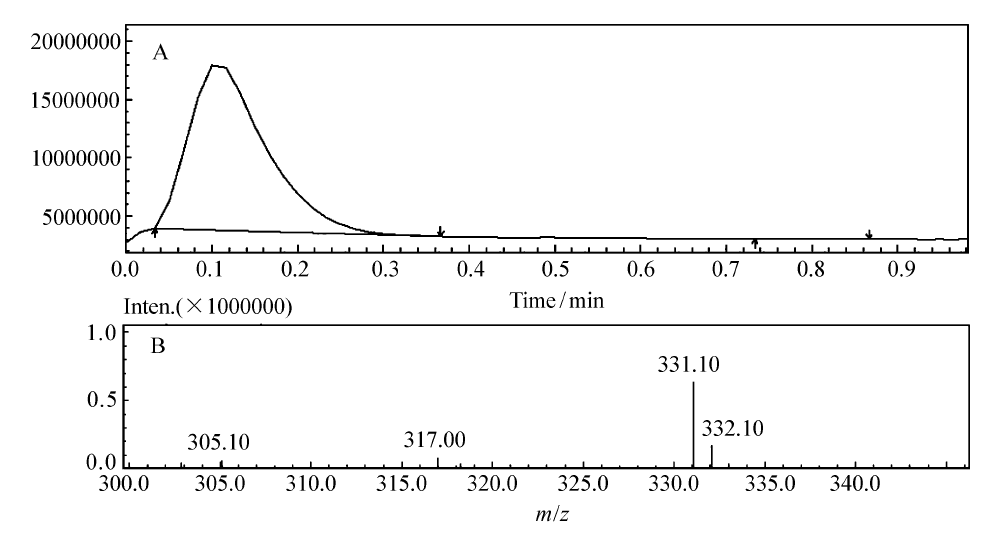 | 图4 2-(芘-1-基)-1,8-萘啶的液相色谱(A)和质谱(B)Fig.4 LC(A) and MS(B) spectra of 2-(pyren-1-yl)-1,8-naphthyridine |
最初拟通过Claisen-Schmidt反应合成含芘查尔酮衍生物3-(2-氨基-吡啶-3-基)-1-(芘-1-基)丙烯酮,但结构表征给出的结果出乎预料,意外地生成了芘联萘啶类化合物2-(芘-1-基)-1,8-萘啶(图5)。 一般来说,氨及其衍生物与芳醛进行缩合反应需在酸催化下进行,酸的作用是增加羰基的反应活性,使其有利于被含氮亲核试剂进攻。 本反应是在碱催化下进行,之所以能够生成2-(芘-1-基)-1,8-萘啶,原因可能在于生成的产物结构是热力学稳定结构。 经讨论分析,认为该反应机理如图6所示(Ar表示芘基)。 首先,在碱作用下发生Claisen-Schmidt缩合反应形成查尔酮中间体。 接着,查尔酮中间体再进行分子内缩合反应,即羰基与氨及其衍生物的加成-消除反应,脱水后形成产物2-(芘-1-基)-1,8-萘啶。
 | 图5 2-(芘-1-基)-1,8-萘啶的合成反应路线图Fig.5 Synthesis of 2-(pyren-1-yl)-1,8-naphthyridine |
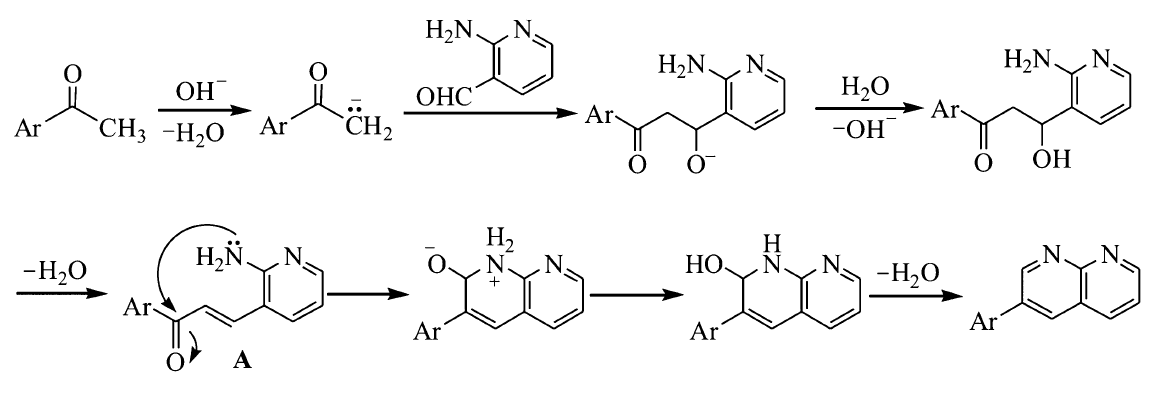 | 图6 2-(芘-1-基)-1,8-萘啶的合成机理示意图Fig.6 Reaction mechanism in the synthesis of 2-(pyren-1-yl)-1,8-naphthyridine |
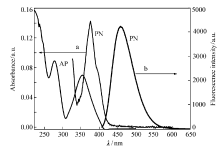
配制浓度为1.0×10-5 mol/L、1×10-6 mol/L的PN的二氯甲烷溶液,分别测定其紫外吸收光谱和荧光发射光谱以及原料乙酰芘AP的紫外吸收光谱(图7)。 由图7可知,PN的最大吸收波长为378 nm,摩尔消光系数1.43×104 L/(mol·cm),与原料AP的紫外最大(强)吸收波长(285 nm)相比,红移93 nm,且在450 nm以上无吸收。 从构效关系看,原料AP和2-氨基吡啶-3-甲醛在碱性条件下缩合生成含芘萘啶结构,形成了更大的 π电子共轭体系,更易发生 π-π*跃迁,使之在紫外光区域有较强的吸收带,并有较大红移。图7显示,PN的荧光最大发射波长为459 nm,荧光强度4240,表现出较强的荧光。
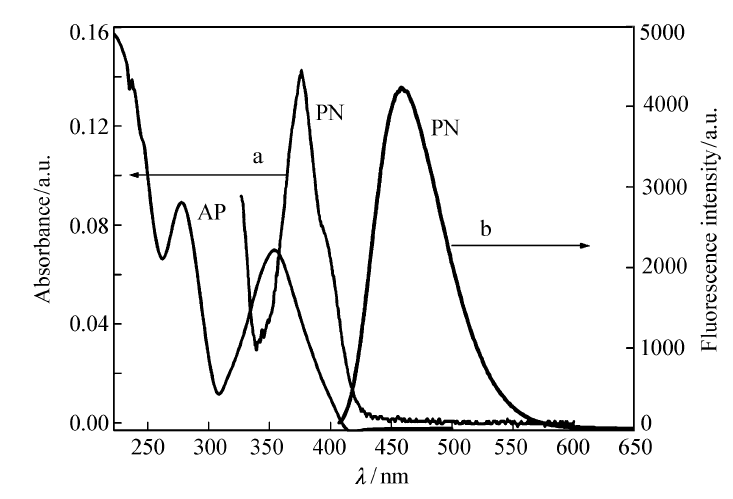 | 图7 2-(芘-1-基)-1,8-萘啶(PN)紫外光谱和1-乙酰基芘(AP)荧光光谱Fig.7 UV(a) and FL(b) spectra of 2-(pyren-1-yl)-1,8-naphthyridine(PN) and UV spectrum of 1-acetylpyrene(AP) |
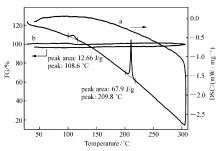
通过热分析仪测定了PN的热学性质,升温速率为10 ℃/min,温度范围为 25~300 ℃,测得的热重/差示扫描量热法(TG/DSC)曲线如图8所示。 PN的熔融峰209.8 ℃,熔融焓22.48 kJ/mol,该熔融峰值与所测熔点209.3~209.8 ℃基本一致。 108.6 ℃处出现1个小吸热峰,熔融焓4.19 kJ/mol,此峰应为不稳定晶型转变的固-固相变峰[14]。 TG曲线基本为一水平线,在25~300℃范围内升温过程中,PN并无明显质量损失,只在晶相转变时有微小热量变化,表现出良好的热稳定性。 这进一步证明,熔融峰209.8 ℃确为化合物的熔化过程,而非分解过程,因为化合物的分解过程一般伴随有质量损失。 综上所述,化合物PN在200 ℃之前不会熔化且无质量损失,可以满足在非线性光学材料应用中的一般要求。
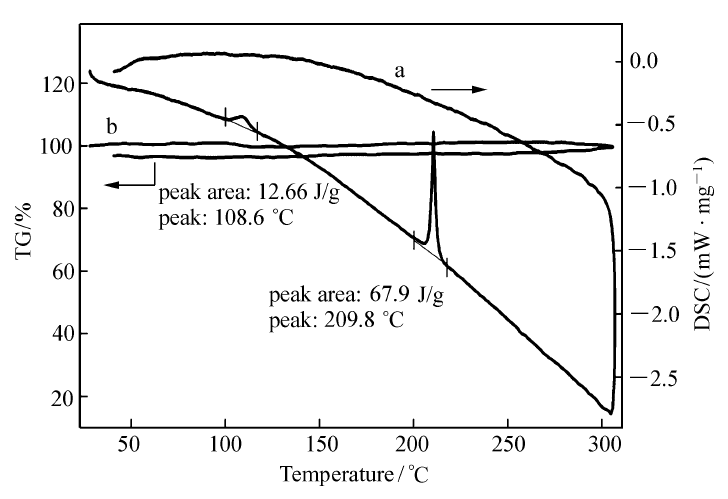 | 图8 2-(芘-1-基)-1,8-萘啶的TG/DSC曲线Fig.8 TG and DSC curves of 2-(pyren-1-yl)-1,8-naphthyridine |
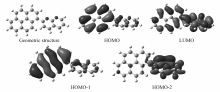
为了探索和理解分子结构和NLO性质之间的构效关系,采用Gaussian09程序[15],对1-(芘-1-基)-1,8-萘啶的几何结构、电子结构和自然轨道(NBO)等在B3LYP/6-311+G(d,p)[16,17]理论水平上进行了计算,图9给出了1-(芘-1-基)-1,8-萘啶分子的几何结构(Geometric structure)、最高占据分子轨道(HOMO)、最低空分子轨道(LUMO)的电子云结构图。
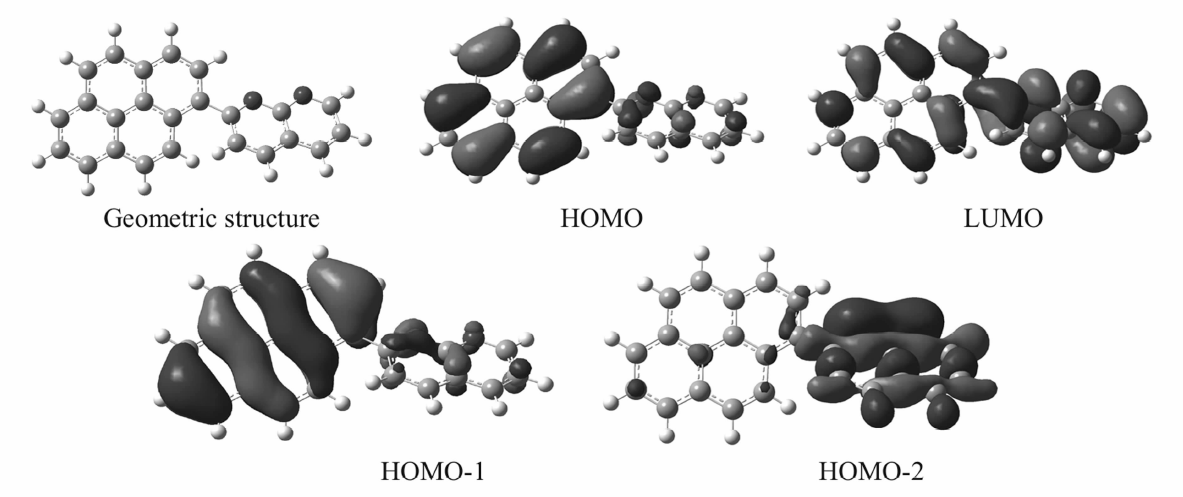 | 图9 2-(芘-1-基)-1,8-萘啶的NBO和分子轨道Fig.9 The NBO and molecular orbitals of 2-(pyren-1-yl)-1,8-naphthyridine |
通过计算分析,获得了PN分子轨道能量和共轭电子体系的极化率和分子超极化率(表1)。
| 表1 2-(芘-1-基)-1,8-萘啶的分子轨道能量、极化率和超极化率 Table 1 Molecular orbital energy, polarization and hyperpolarization of 2-(pyren-1-yl)-1,8-naphthyridine |
由表1可知,1-(芘-1-基)-1,8-萘啶分子的LUMO与HOMO两轨道之间的能级差较小,化合物分子有较大的极化率和超极化率,意味着分子内更容易发生电荷转移。图8显示,电子在 π-π*跃迁过程中的电子云变化极为明显。 可以看到,LUMO与HOMO两轨道的电子云从分子的一端跃迁到另一端的电荷转移比较明显,而HOMO-1与HOMO-2两轨道的这种变化更加明显。图9中HOMO显示1-(芘-1-基)-1,8-萘啶分子中电子云主要集中于芘基,同时LUMO显示该分子中相当一部分电子云转移到了萘啶基。 显然,芘基为电子给体,萘啶基则为电子受体(HOMO-1与HOMO-2两轨道的这种变化进一步证明了这一点)。 从构效关系来说,电子给体与电子受体直接连在一起,形成D-A型的共轭体系,意味着分子更易被极化。 计算所得能隙、极化率和超极化率数据也表明,化合物分子中氮原子的强拉电子作用是 π电子体系极化以及分子内电荷分离变化的根源。
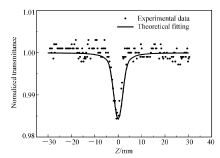
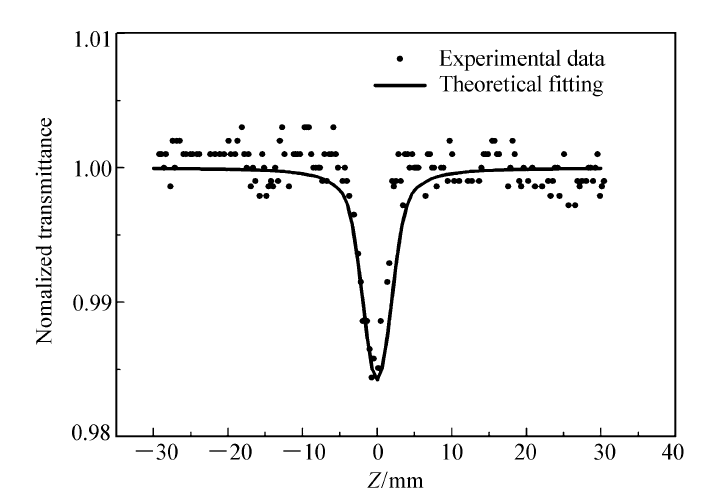
采用Z-扫描技术测定,选用可调谐激光器作为光源,激光脉冲180 fs,波长532 nm,样品浓度5.78 mmol/L的二甲基亚砜溶液(DMSO),重复频率10 Hz,实验获得了1-(芘-1-基)-1,8-萘啶的三阶非线性光学吸收曲线图(图10),通过数值拟合获得三阶非线性光学吸收系数 β=9.0×10-14 m/W。 1-(芘-1-基)-1,8-萘啶在上述条件下表现出反饱和吸收( β>0);在吸收过程中化合物表现出超快响应,意味着它的非线性响应的灵敏度较高;吸收系数与极化率具有一致性,即吸收系数大时对应的极化率也大。 从构效关系看,两个吡啶环相并形成的萘啶基作为电子受体和芘基相连形成了D-A型共轭结构,更有利于 π电子极化从而增大其反饱和吸收系数。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
合成了一个新的NLO有机材料2-(芘-1-基)-1,8-萘啶,该化合物分子属于芘联萘啶类D-A型共轭结构。 理论计算和非线性性质测定表明,这种特有的D-A型共轭结构使得该化合物分子易于发生电荷转移从而增大其极化率并在飞秒激光条件下呈现出反饱和吸收现象。 该化合物的紫外光谱在450 nm以上无吸收,具有良好的热稳定性,可以在非线性光学吸收、激光防护、吸收型光开关或双稳器件等方面用作备选材料。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|