

GUO Hongchen, QIN Yusheng, WANG Xianhong, et al. Copolymerization of Carbon Dioxide and Propylene Oxide under Aluminum Porphyrin Catalyst[J]. Chinese Journal of Applied Chemistry, 36(10): 1118-1127


铝卟啉是一类土壤环境友好的金属卟啉,尽管早在1978年Inoue就已经发现它可以催化CO2和环氧丙烷的共聚反应,但是该催化体系一直面临催化活性低、聚合物相对分子质量低等难题。 本文通过改变铝卟啉催化剂配体中苯环上取代基的种类和位置,制备出中心金属电子环境差异化的铝卟啉,并以双三苯基膦氯化铵(PPNCl)为助催化剂,探讨其对CO2与环氧丙烷的共聚反应的催化行为。 结果表明,当铝卟啉中苯环上2,4位同时被Cl-取代后,在90 ℃和3 MPa压力下,转化频率(TOF)达到2672 h-1。 当利用离去能力较强的对甲苯磺酸基团(OTs-)作为铝卟啉的轴向配体,可以合成出数均相对分子质量达1.84×105的脂肪族聚碳酸酯。
Aluminum porphyrin is a soil-tolerant metal porphyrin complex. Although its catalytic activity on the copolymerization of CO2 and propylene oxide has been disclosed by Inoue in 1978, the catalytic activity is still very low, and the synthesized poly(propylene carbonate) has low relative molecular mass. It is a big challenge to make progress on the catalytic performance of aluminum porphyrin. In this work, the electronic environment of central aluminum was adjusted by delicate design of porphyrin ligand using meso-tetrasubstituted porphyrin derivatives that were employed to catalyze the copolymerization of CO2 and propylene oxide with bis-(triphenyl phosphine) iminium chloride(PPNCl) as the co-catalyst. It was found that the electronic environment of the central aluminum ion had great effect on the catalytic performance of aluminum porphyrin catalysts, the turnover frequency(TOF) value of Cl substituted aluminum porphyrin catalyst 6a reached 2672 h-1 at 90 ℃ and 3 MPa, while poly(propylene carbonate) with relative molecular mass of 1.84×104 was afforded using catalyst 4b bearing toluene sulfonic group(OTs-) as axial group of good leaving ability. Our work indicates that delicate designed aluminum porphyrin can become a possible candidate as high performance catalyst in the copolymerization of CO2 and propylene oxide, under optimized copolymerization conditions.
二氧化碳(CO2)是一种来源丰富、价格低廉的C1资源,以其为原料制备成化工产品是一条利用CO2的有效途径[1,2],尤其是利用CO2与环氧化物反应合成环状碳酸酯[3,4]或聚碳酸酯[5,6,7,8],已经成为高附加值利用CO2的一个发展趋势。 由于CO2本身在热力学上高度稳定,必须设计合适的催化剂以大幅度降低其相关反应的活化能,从而实现CO2的快速活化和高效转化[9]。
1969年,Inoue等[10]首次利用二乙基锌/水(ZnEt2/H2O)催化CO2与环氧丙烷(PO)反应合成了聚碳酸丙烯酯(PPC),在随后的近50年内研究人员发展出许多高效催化体系,其中以稀土三元催化剂为代表的非均相催化剂下PPC的制造已经实现了万吨级工业化,但是其催化活性依然亟待提高,而且催化剂的选择性、聚合物相对分子质量的调整依然是该领域的难题[6,8]。 在均相催化体系中,Salen钴配合物催化体系无论是用于CO2与环氧化物的偶联反应还是共聚反应,均表现出很高的活性和选择性,是该领域综合性能最好的催化剂体系[11]。 该催化体系始于2003年Coates等[12]制备的SalenCoX催化剂,随后经历了Nozaki[13]、Lu[11]、Lee[14]等对配体取代基和轴向基团的巧妙设计,获得的单组元双功能SalenCoX催化剂的转化频率(TOF)可达26000 h-1,并制备出相对分子质量高达3.74×105的PPC,且在较高温度或极低催化剂浓度下仍能保持很好的催化性能[14]。 不过中心金属钴为重金属,而生物降解塑料严格限制了钴的残留,因此很大程度上限制了其应用[15]。
中心金属为铝的配合物对土壤的危害可以忽略不记,是一类土壤环境友好的催化体系[16]。 尽管在1978年Inoue等[17]就发现铝卟啉可以催化CO2与环氧丙烷的共聚反应,但是其催化活性很低,所得PPC的相对分子质量也很低。 Ree等[18]以四苯基卟啉氯化铝(TPPAlCl)为催化剂、四乙基溴化铵为助催化剂,制备了碳酸酯链段含量为70%~75%的PPC,相对分子质量仅为1900~3300。 我们通过大位阻Lewis酸的加入,有效地提高了TPPAlCl的催化活性[19],但与已有的Salen金属催化剂相比,铝卟啉在催化性能方面仍存在很多不足。 受双官能Salen催化剂的启发,我们在卟啉配体上键接季铵盐并调节中心铝的电子环境,在80 ℃和3 MPa下TOF达560 h-1,PPC选择性达到93%,所制备的PPC数均相对分子质量达9.6×104[20],不过键接季铵盐操作复杂,成本高,难以大规模应用。 Coates等[21]提出通过调节配体的电子效应可以改变中心金属的Lewis酸性,进而提高催化活性。
本文以卟啉铝为主催化剂,以双三苯基膦氯化铵(PPNCl)为助催化剂,通过调整卟啉催化剂苯环上取代基团的种类和位置以及轴向基团,调节中心金属铝的电子环境,试图提高催化活性,同时提高PPC的选择性和相对分子质量。
苯甲醛(≥99.5%)、4-氯苯甲醛(≥98%)、4-氟苯甲醛(98%)、4-溴苯甲醛(≥99%)、2-氯苯甲醛(≥98%)、2,4-二氯苯甲醛(≥98%)、2,4,6-三氯苯甲醛(≥98%)、对甲苯磺酸银(≥98%)和吡咯(≥99%)均购自阿拉丁试剂有限公司,使用前,吡咯经蒸馏(Ar气保护)后待用;三氟乙酸(≥99%)、二氯二氰基苯醌(DDQ)(≥99%)、三氟甲基磺酸银(≥99%)、双三苯基膦氯化铵(PPNCl,≥97%)购自美国Sigma-Aldrich公司;中性氧化铝(48~75 μm),分析纯,购自上海国药试剂有限公司;环氧丙烷(≥99.7%)购自吉神化学工业股份有限公司,CaH2蒸馏后使用;CO2(≥99.95%)购自四平健新气体有限公司,直接使用。二氯甲烷,分析纯,购自北京化工厂,CaH2蒸馏(Ar气保护)后使用。
Bruker ARX-300型核磁共振谱仪(NMR,德国布鲁克公司);Waters 410型凝胶渗透色谱仪(GPC,美国沃特世公司),色谱柱为Waters HT-4、HT-5两柱串联,标样为聚苯乙烯,二氯甲烷为流动相,柱温35 ℃,流速为1 mL/min。
1.2.1 卟啉配体的合成
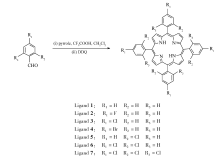
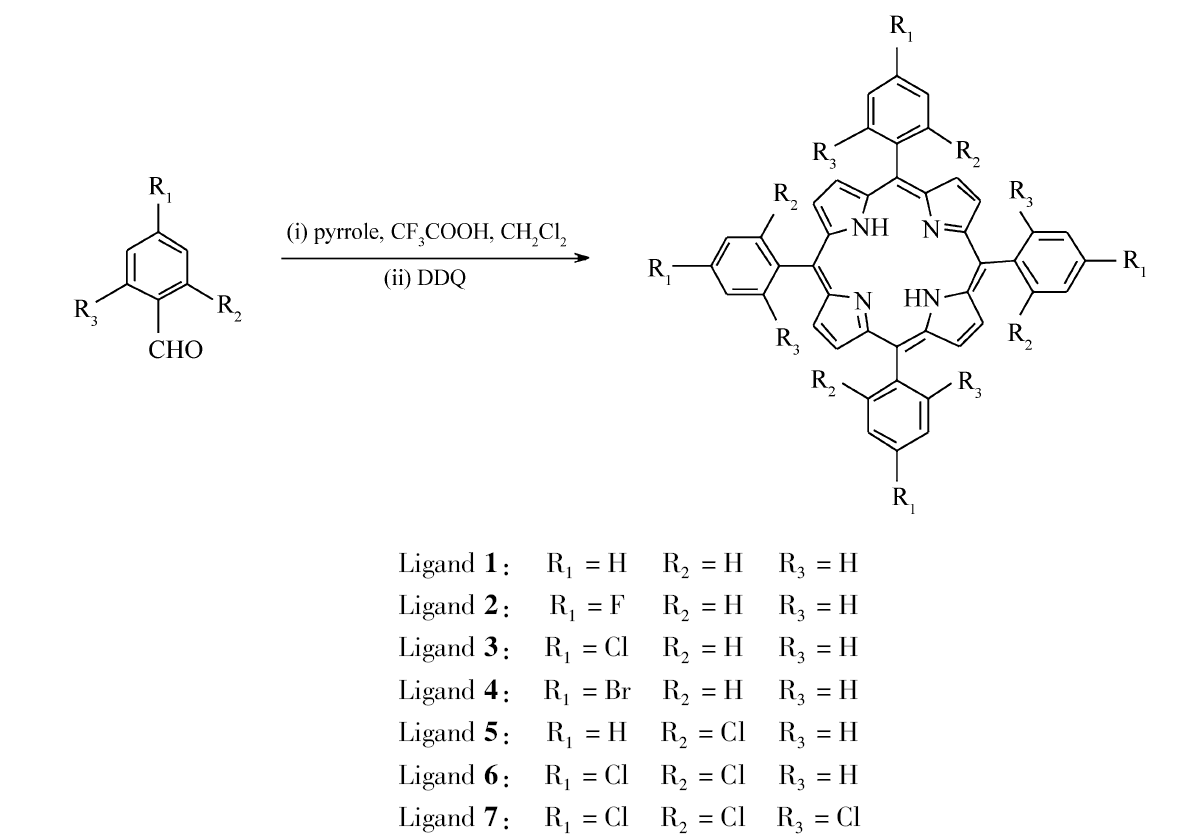
配体1的合成 Ar气保护下将0.7 mL吡咯和1.06 g苯甲醛加入到预先装有400 mL二氯甲烷的500 mL圆底三口瓶中,随后加入0.7 mL三氟乙酸,25 ℃下搅拌1 h后加入4.54 g DDQ,搅拌1 h后过滤,滤液采用减压蒸馏除去二氯甲烷得到紫黑色固体,经色谱柱分离(固定相:中性三氧化二铝;流动相: V(二氯甲烷): V(石油醚)=1:1),产物收率9.1%。
1H NMR(CDCl3,300 MHz), δ:8.89(s,8H),8.15(m,8H),7.81(m,12H),-2.72(s,2H);13C NMR(CDCl3,100 MHz), δ:142.2,134.6,131.0,127.7,126.7,120.1。
配体2的合成 在Ar气保护下,将0.7 mL吡咯和1.24 g对氟苯甲醛加入到预先装有400 mL二氯甲烷的500 mL圆底三口瓶中,随后加入0.7 mL三氟乙酸,25 ℃下搅拌1 h后加入4.54 g DDQ,搅拌1 h后过滤,滤液采用减压蒸馏除去二氯甲烷得到紫黑色固体,经色谱柱分离(固定相:中性三氧化二铝;流动相: V(二氯甲烷): V(石油醚)=1:1),产物收率10.3%。
1H NMR(CDCl3,400 MHz), δ:8.83(s,8H),8.17(d,8H),7.48(d,8H),-2.83(s,2H);13C NMR(CF3COOD,100 MHz), δ:134.8,129.0,122.4,118.3,112.7,109.9。
配体3的合成 在Ar气保护下,将0.7 mL蒸过的吡咯和1.41 g对氯苯甲醛加入到装有400 mL蒸过的二氯甲烷的500 mL圆底三口瓶中,随后加入0.7 mL三氟乙酸,25 ℃下搅拌1 h后加入4.54 g DDQ,搅拌1 h后过滤,滤液采用减压蒸馏除去二氯甲烷得到紫黑色固体,经色谱柱分离(固定相:中性三氧化二铝;流动相: V(二氯甲烷): V(石油醚)=1:1),产物收率13.5%。
1H NMR (CDCl3,300 MHz), δ:8.84(s,8H),8.12(d,8H),7.76(d,8H),-2.86(s,2H);13C NMR (CDCl3,100 MHz), δ:140.3,135.5,134.4,131.1,127.0,118.9。
配体4的合成 Ar气保护下将0.7 mL吡咯和1.85 g对溴苯甲醛加入到装有400 mL蒸过的二氯甲烷的500 mL圆底三口瓶中,随后加入0.7 mL三氟乙酸,25 ℃下搅拌1 h后加入4.54 g DDQ,搅拌1 h后过滤,滤液采用减压蒸馏除去二氯甲烷得到紫黑色固体,经色谱柱分离(固定相:中性三氧化二铝;流动相: V(二氯甲烷): V(石油醚)=1:1),产物收率14.5%。
1H NMR(CDCl3,300 MHz), δ:8.84(s,8H),8.06(m,8H),7.88(m,8H),-2.86(s,2H);13C NMR(CDCl3,100 MHz), δ:140.8,135.8,131.9,122.6,119.0。
配体5的合成 Ar气保护下将0.7 mL吡咯和1.41 g 2-氯苯甲醛加入到装有400 mL蒸过的二氯甲烷的500 mL圆底三口瓶中,随后加入0.7 mL三氟乙酸,25 ℃下搅拌1 h后加入4.54 g DDQ,搅拌1 h后过滤,滤液采用减压蒸馏除去二氯甲烷得到紫黑色固体,经色谱柱分离(固定相:中性三氧化二铝;流动相: V(二氯甲烷): V(石油醚)=1:1),产物收率11.5%。
1H NMR(CDCl3,300 MHz), δ:8.83(s,8H),8.20(m,4H),7.74(m,12H),-2.64(s,2H);13C NMR(CDCl3,100 MHz), δ:140.8,135.7,131.1,125.6,117.1。
配体6的合成 Ar气保护下将0.7 mL吡咯和1.75 g 2,4-二氯苯甲醛加入到装有400 mL二氯甲烷的500 mL圆底三口瓶中,随后加入0.7 mL三氟乙酸,25 ℃下搅拌1 h后加入4.54 g DDQ,搅拌1 h后过滤,滤液采用减压蒸馏除去二氯甲烷得到紫黑色固体,经色谱柱分离(固定相:中性三氧化二铝;流动相: V(二氯甲烷): V(石油醚)=1:1),产物收率10.5%。
1H NMR(CDCl3,300 MHz), δ:8.71(s,8H),8.05(m,4H),7.92(m,4H),7.55(m,4H),-2.74(s,2H);13C NMR(CDCl3,100 MHz), δ:139.2,137.4,135.8,135.2,130.6,129.3,126.1,116.1。
配体7的合成 在Ar气保护下,将0.7 mL蒸过的吡咯和2.09 g 2,4,6-三氯苯甲醛加入到装有400 mL蒸过的二氯甲烷的500 mL圆底三口瓶中,随后加入0.7 mL三氟乙酸,25 ℃下搅拌1 h后加入4.54 g DDQ,搅拌1 h后过滤,滤液采用减压蒸馏装置除去二氯甲烷得到紫黑色固体,经色谱柱分离(固定相:中性三氧化二铝;流动相: V(二氯甲烷): V(石油醚)=1:1),产物收率3.1%。
1H NMR(CDCl3,400 MHz), δ:8.66(s,8H),7.87(s,8H),-2.63(s,2H);13C NMR(CDCl3,100 MHz), δ:161.1,139.5,138.3,136.1,128.3,127.3,113.8。
卟啉配体1-7的合成示意图如Scheme 1所示。
 | scheme 1 Synthesis route to porphyrin ligands 1-7 |
{kind=link}
1.2.2 金属卟啉铝配合物的合成
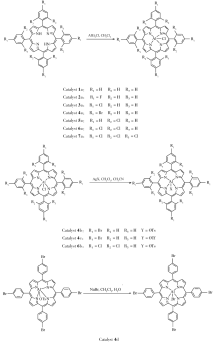
催化剂1a的合成:Ar气保护下将20 mL二氯甲烷加入到装有0.615 g配体1的50 mL圆底三口瓶中,25 ℃下搅拌至配体完全溶解,随后加入1.3 mL二乙基氯化铝的正己烷溶液(1 mol/L),25 ℃下搅拌1 h后使用旋转蒸发仪旋干溶剂,粗产物经色谱柱分离(固定相:中性三氧化二铝;流动相: V(二氯甲烷): V(甲醇)=10:1),产物收率98.5%。
1H NMR(CDCl3,300 MHz), δ:8.79(s,8H),8.17(m,8H),7.82(m,12H);13C NMR(CDCl3,100 MHz), δ:146.4,142.1,134.0,132.0,127.6,126.9,119.5。
催化剂2a的合成:Ar气保护下将20 mL二氯甲烷加入到装有0.687 g配体2的50 mL圆底三口瓶中,25 ℃下搅拌至配体完全溶解,随后加入1.3 mL二乙基氯化铝的正己烷溶液(1 mol/L),25 ℃下搅拌1 h后使用旋转蒸发仪旋干溶剂,粗产物经色谱柱分离(固定相:中性三氧化二铝;流动相: V(二氯甲烷): V(甲醇)=10:1),产物收率98.0%。
1H NMR(CDCl3,300 MHz), δ:9.04(s,8H),8.25(m,8H),7.98(m,8H);13C NMR(CDCl3,100 MHz), δ:146.7,139.3,135.5,131.8,129.0,126.6,117.1。
催化剂3a的合成 Ar气保护下将20 mL二氯甲烷加入到装有0.753 g配体3的50 mL圆底三口瓶中,25 ℃下搅拌至配体完全溶解,随后加入1.3 mL二乙基氯化铝的正己烷溶液(1 mol/L),25 ℃下搅拌1 h后使用旋转蒸发仪旋干溶剂,粗产物经色谱柱分离(固定相:中性三氧化二铝;流动相: V(二氯甲烷): V(甲醇)=10:1),产物收率98.5%。
1H NMR(CDCl3,300 MHz), δ:9.04(s,8H),8.25(m,8H),7.98(m,8H);13C NMR(CDCl3,100 MHz), δ:146.5,139.6,135.5,133.5,132.2,127.1,119.0。
催化剂4a的合成 Ar气保护下将20 mL二氯甲烷加入到装有0.930 g配体3的50 mL圆底三口瓶中,25 ℃下搅拌至配体完全溶解,随后加入1.3 mL二乙基氯化铝的正己烷溶液(1 mol/L),25 ℃下搅拌1 h后使用旋转蒸发仪旋干溶剂,粗产物经色谱柱分离(固定相:中性三氧化二铝;流动相: V(二氯甲烷): V(甲醇)=10:1),产物收率98.1%。
1H NMR(CDCl3,300 MHz), δ:9.03(s,8H),8.15(m,8H),8.06(m,8H);13C NMR(CDCl3,100 MHz), δ:146.4,140.0,135.8,132.2,130.0,122.2,119.0。
催化剂5a的合成 Ar气保护下将20 mL二氯甲烷加入到装有0.753 g配体3的50 mL圆底三口瓶中,25 ℃下搅拌至配体完全溶解,随后加入1.3 mL二乙基氯化铝的正己烷溶液(1 mol/L),25 ℃下搅拌1 h后使用旋转蒸发仪旋干溶剂,粗产物经色谱柱分离(固定相:中性三氧化二铝;流动相: V(二氯甲烷): V(甲醇)=10:1),产物收率98.0%。
1H NMR(CDCl3,300 MHz), δ:8.92(s,8H),8.31(m,4H),7.92(m,12H);13C NMR(CDCl3,100 MHz), δ:163.6,161.1,146.7,137.1,135.6,132.1,119.1,114.0。
催化剂6a的合成 Ar气保护下将20 mL二氯甲烷加入到装有0.890 g配体3的50 mL圆底三口瓶中,25 ℃下搅拌至配体完全溶解,随后加入1.3 mL二乙基氯化铝的正己烷溶液(1 mol/L),25 ℃下搅拌1 h后使用旋转蒸发仪旋干溶剂,粗产物经色谱柱分离(固定相:中性三氧化二铝;流动相: V(二氯甲烷): V(甲醇)=10:1),产物收率98.6%。
1H NMR(CDCl3,300 MHz), δ:8.95(s,8H),8.28(m,8H),7.92(m,4H);13C NMR(CDCl3,100 MHz), δ:146.6,138.1,136.4,134.8,132.0,132.0,128.6,126.4,116.0。
催化剂7a的合成 Ar气保护下将20 mL二氯甲烷加入到装有1.028 g配体3的50 mL圆底三口瓶中,25 ℃下搅拌至配体完全溶解,随后加入1.3 mL二乙基氯化铝的正己烷溶液(1 mol/L),25 ℃下搅拌1 h后使用旋转蒸发仪旋干溶剂,粗产物经色谱柱分离(固定相:中性三氧化二铝;流动相: V(二氯甲烷): V(甲醇)=10:1),产物收率99.1%。
1H NMR(CDCl3,400 MHz),δ:8.91(s,8H),8.42(s,8H);13C NMR(CDCl3,100 MHz), δ:145.2,141.1,134.1,131.3,128.1,123.8,117.8。
催化剂4b的合成 Ar气保护下将0.991 g催化剂4a加入到装有50 mL二氯甲烷的100 mL圆底烧瓶中,25 ℃下搅拌溶解,随后加入20 mL溶有0.419 g对甲苯磺酸银的乙腈溶液,25 ℃下搅拌12 h后使用旋转蒸发仪旋干溶剂,粗产物经过二氯甲烷溶解后过滤,滤液使用旋转蒸发仪除去溶剂,产物收率98.5%。
1H NMR(CDCl3,300 MHz), δ:9.03(s,8H),8.14(d,8H),8.05(s,8H),7.46(s,2H),7.10(d,2H),2.28(s,3H);13C NMR(CDCl3,100 MHz), δ:146H5,142.0,135.8,132.3,130.1,128.0,125.5,118.3,108.6,20.8。
催化剂4c的合成 在Ar气保护下,将0.991 g催化剂4a加入到装有50 mL二氯甲烷的100 mL圆底烧瓶中,25 ℃下搅拌溶解,随后加入20 mL溶有0.386 g对三氟甲磺酸银的乙腈溶液,25 ℃下搅拌12 h后使用旋转蒸发仪旋干溶剂,粗产物经过二氯甲烷溶解后过滤,滤液使用旋转蒸发仪除去溶剂,产物收率98.0%。
1H NMR(CDCl3,400 MHz), δ:9.05(s,8H),8.15(d,8H),8.06(s,8H);13C NMR(CDCl3,100 MHz), δ:146.5,146.1,140.9,140.0,135.8,132.3,131.2,130.0,129.6,122.2,121.7,119.0,118.2,35.2。
催化剂6b的合成 在Ar气保护下,将0.951 g催化剂4a加入到装有50 mL二氯甲烷的100 mL圆底烧瓶中,25 ℃下搅拌溶解,随后加入20 mL溶有0.419 g对甲苯磺酸银的乙腈溶液,25 ℃下搅拌12 h后使用旋转蒸发仪旋干溶剂,粗产物经过二氯甲烷溶解后过滤,滤液使用旋转蒸发仪除去溶剂,产物收率98.8%。
1H NMR(CDCl3,400 MHz), δ:8.95(s,8H),8.28(m,8H),7.95(m,4H),7.46(d,2H),7.10(d,2H),2.28(s,3H);13C NMR(CDCl3,100 MHz), δ:146.6,145.4,138.2,137.5,136.5,134.8,132.0,128.5,128.0,126.4,125.4,116.0,20.7。
催化剂4d的合成 Ar气保护下将1.126 g催化剂4b加入到装有50 mL二氯甲烷的100 mL圆底烧瓶中,25 ℃下搅拌溶解,随后加入40 mL溶有0.419 g溴化钠溶液,25 ℃下搅拌2 h后使用分液漏斗收集有机相,用水洗3次后,有机相用无水硫酸钠干燥,过滤,用旋转蒸发仪除去溶剂,产物收率98.5%。
1H NMR(CDCl3,300 MHz), δ:9.02(s,8H),8.13(m,8H),8.06(m,8H);13C NMR(CDCl3,100 MHz), δ:146.4,140.0,135.7,132.3,129.9,121.7,118.2。
卟啉催化剂1-7的合成示意图如Scheme 2所示。
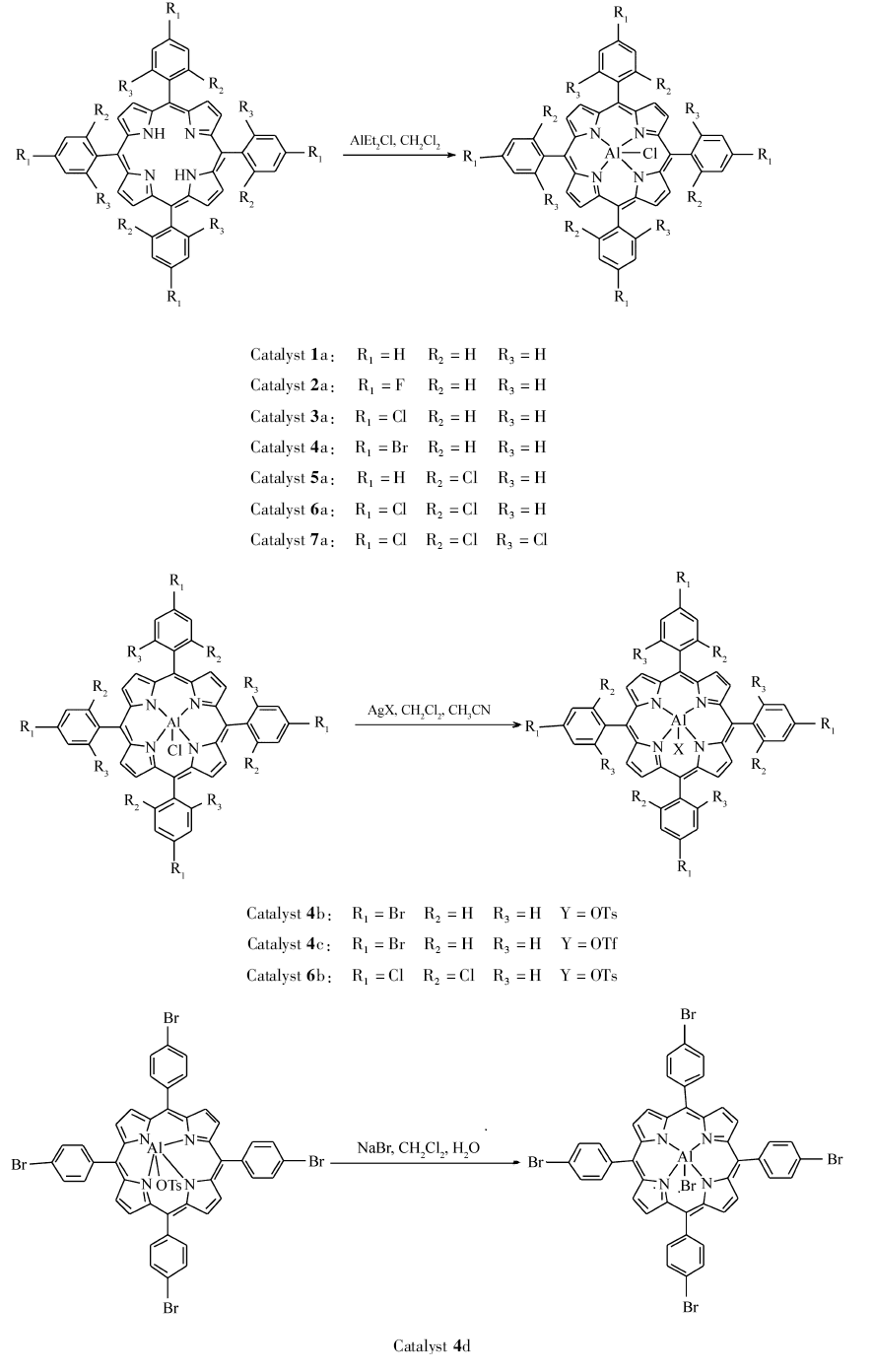 | scheme 2 Synthesis route to porphyrin catalysts 1-7 |
{kind=link}
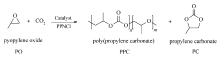
将上述制备的催化剂及助催化剂PPNCl加入到预先除水、除氧处理的高压反应釜内,加入一定比例的PO,通入CO2,在3 MPa和温度下搅拌,反应一定时间后,降温终止反应,室温下排出CO2气体,减压除去未反应的PO,取适量反应产物进行1H NMR和GPC分析,通过核磁与产物最终质量计算催化剂的TOF值。 CO2与PO的共聚反应简图如Scheme 3所示。
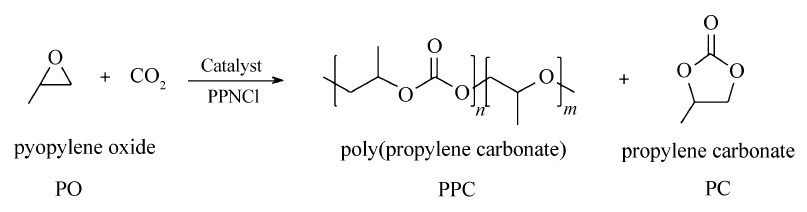 | scheme 3 Copolymerization of epoxide and carbon dioxide |
{kind=link}
催化剂1a-7a对CO2与PO共聚反应的催化性能如表1所示。 铝卟啉配体中苯环对位分别被氟、氯、溴取代后(Entry 2-4),催化活性由235提高到310 h-1。 其中对位被氯取代后PPC的选择性最高(96.1%),所得PPC的碳酸酯含量97.8%,数均相对分子质量( Mn)2.67×104,与四苯基卟啉氯化铝相比催化性能有所提高,说明配体苯环上引入吸电子基团后改善了中心金属的电子环境,进而影响铝卟啉的催化性能。
| 表1 带有不同取代基的铝卟啉的催化性能 Table 1 Copolymerization of CO2/PO catalyzed by catalyst 1a-7a a |
根据表1的Entry 5-7,当苯环的邻位被取代时,碳酸酯含量均大于99.9%,主要原因是具有吸电子能力的氯原子键接在苯环邻位后,离中心金属更近,使中心金属具有更高的Lewis酸性,增强了阴离子增长链与中心金属之间的作用力,降低了烷氧基开环PO的速率,增加了CO2的插入几率,进而提高了PPC的碳酸酯含量。 铝卟啉配体中苯环上2,4位同时被氯取代后,催化活性最高,达到582 h-1,并且 Mn提高到4.58×104,不过PPC的选择性则下降到92.5%。
以催化活性较高的催化剂6a为代表,以PPNCl为助催化剂,在70 ℃、3 MPa下聚合反应3 h,共聚反应结果列于表2。 随着助催化剂PPNCl的比例从催化剂的10%提高到200%,催化剂6a的TOF从217提高到770 h-1,PPC的选择性则逐渐降低。 当助催化剂是催化剂的200%(2倍)时PPC的选择性低于90%,但是PPC中的碳酸酯含量不随助催化剂与催化剂的比例变化而变化,均保持在很高的水平(99.9%),聚合物相对分子质量则先随助催化剂与催化剂比例的提高而增加,当 n(Al): n(PPNCl)=1:1时,聚合物 Mn最高,达到4.58×104,之后 Mn逐渐下降,因此 n(Al): n(PPNCl)=1:1时,综合催化性能最佳。
| 表2 不同助催化剂用量下铝卟啉的催化性能 Table 2 Influence of co-catalyst/catalyst ratio on copolymerization of CO2/PO a |
选取催化剂4a为研究对象,用Br-、对甲苯磺酸基团(OTs-)、三氟甲磺酰基(OTf-)代替原有的轴向Cl-离子,以PPNCl为助催化剂,在70 ℃、3 MPa下聚合3 h,PO与CO2共聚反应的结果列于表3。 根据表3的Entry 1-4数据,轴向基团的离去能力按如下顺序逐步增强:Cl-、Br-、OTs-、OTf-,相应地,铝卟啉的催化活性逐渐降低,但聚合物选择性和聚合物的 Mn均是先增后减,轴向离子为OTs-时铝卟啉的综合催化活性最好,说明轴向基团对中心金属的电子环境影响较大,随着轴向基团离去能力的增强,在抑制催化活性的同时,聚合物的选择性却得到了大幅提高,聚合物的 Mn也有了相应增长。 但当轴向基团的离去能力过强时,只能抑制催化活性,聚合物的选择性和聚合物 Mn均无提高。
| 表3 不同轴向基团金属卟啉铝催化剂的催化性能 Table 3 Copolymerization of CO2/PO catalyzed by catalysts 4a-4d, 6a, 6b a |
如前所述,通过调整催化剂4a的轴向基团,可以使聚合物的选择性和 Mn得到提高。为此将催化活性较高的催化剂6a的轴向基团改为OTs-基团(催化剂6b,Entry 6),却发现活性大幅下降,从582降低到117 h-1,聚合物的选择性只提高了2.1%, Mn也因为转化率较低而降低了,可见轴向基团的选择需要非常谨慎,目前还是需要试错模式。
以催化剂4b和6a为主催化剂,研究了温度对PO与CO2共聚反应的影响,结果分别列于表4的Entry 1-4和Entry 8-10。 随着反应温度的上升,催化活性大幅提高,其中催化剂6a在90 ℃时TOF达到2672 h-1,聚合物碳酸酯含量99.9%,但聚合物选择性只有74.6%,对应聚合物的 Mn也只有3.90×104。 不过催化剂4b由于有轴向OTs-的影响, 100 ℃时,聚合物的选择性仍有89.0%,聚合物选择性95.8%,TOF达到1044 h-1,聚合物的 Mn也有7.42×104。 综合温度对PO和CO2共聚合的影响,选择催化剂4b为主催化剂,在80 ℃下,调整反应时间和催化剂的浓度,得到Entry 7所示的结果,聚合物选择性93.2%,碳酸酯含量95.8%, Mn高达1.84×105的PPC。 值得指出的是,表4中所示的聚合物相对分子质量分布均小于1.20。
| 表4 反应条件对铝卟啉催化性能的影响 Table 4 Copolymerization of CO2/PO catalyzed by catalysts 4b, 6a a |
通过改变卟啉配体苯环上取代基的种类和位置,制备了中心金属电子环境不同的铝卟啉催化剂作为主催化剂,与双三苯基膦氯化铵(PPNCl)助催化剂相结合,用于催化CO2与环氧丙烷(PO)的共聚反应。 当铝卟啉配体苯环邻位上的氢被取代后,可有效地提高聚合物的碳酸酯含量,如催化剂5a-7a以及6b催化得到的聚合物碳酸酯含量高达99.9%。 当铝卟啉配体苯环上2,4号位被氯取代后,所形成的催化剂6a的催化活性最高,在90 ℃,3 MPa下,TOF达到2672 h-1。 利用离去能力较强的对甲苯磺酸基团(OTs-)取代Cl-作为轴向基团制备了催化剂4b,在80 ℃和3 MPa下,可以制备 Mn达到1.84×105的PPC。 铝卟啉催化剂不仅具有环境友好的中心金属,还具有高催化活性、高PPC选择性和生成高相对分子质量PPC的3个特点,有望成为CO2与PO共聚领域很有潜力的均相催化剂。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|