

ZHOU Liwang, ZHAO Zhouxing. Synthesis of Aryl Formic Acid Cyano Aryl Methyl Esters[J]. Chinese Journal of Applied Chemistry, 36(1): 10-15


芳甲酸氰基芳甲酯是重要的有机合成中间体,其现有合成方法采用剧毒氰化物为氰源来合成。 本研究以K4[Fe(CN)6]为绿色氰化试剂,芳酰氯为原料,采用一锅两步反应合成芳甲酸氰基芳甲酯。 通过改变第二步反应温度、反应时间、硼氢化钠和催化剂的用量获得最佳反应条件,以61.7%~80.3%的产率合成了10种芳甲酸氰基芳甲酯(2a~2j),产物结构通过傅里叶变换红外光谱仪(FTIR)、核磁共振波谱仪(NMR)分析确认。 根据实验结果,提出了可能的反应机理。 该法避免了对剧毒氰化剂的使用,具有产率高、操作简单、后处理方便等优点。
Aryl formic acid cyano aryl methyl esters are important organic synthesis intermediate presently obtained from highly toxic cyanide. In this study, K4[Fe(CN)6] as a green cyanide source combined with acyl chlorine was used to synthesize this type of cyanohydrin ester in a one pot two-steps reaction. Optimized reaction condition was obtained by changing the temperature in second step, the reaction time, the dosages of sodium borohydride and the catalysts,Ten aryl formic acid cyano aryl methyl esters(2a~2j) were synthesized in 61.7%~80.3% yield. Their structures were confirmed by Fourier transform infrared spectrometer(FTIR) and nuclear magnetic resonance spectrometer(NMR). A possible reaction mechanism was also proposed. This method avoids the use of highly toxic cyanide, and benefits from high yield, simple operation, and convenient post-processing.
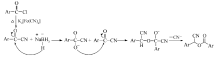
芳甲酸氰基芳甲酯是重要的有机合成中间体。 在农药、染料和医药等领域有广泛的应用[1,2,3]。 其常见的合成方法有碱催化下芳酰基腈自身偶联[4,5,6]、三甲基硅氰(TMS-CN)或金属氰化剂与醛酮的反应[7,8]、芳基氰化物的还原耦合[9]和酰氯与氰醇的反应[10]等。 这些合成法中常用金属氰化物(NaCN、KCN)、TMS-CN、芳酰基腈、氰醇等为氰源。 而金属氰化物是剧毒物质,TMS-CN对湿空气极其敏感,容易吸潮而放出有毒的氰化氢气体。 另外,市售酰基腈和氰醇化合物不仅价格昂贵,种类数量也极其有限,并且也是由商业上可得的剧毒氰化物制备而得,所以,这些氰化剂的使用极不安全,同时对环境还会造成污染。
K4[Fe(CN)6]是一种安全无毒含氰化合物,是煤化学工业的副产品,产量很大,价格低廉,在食品工业中作为金属沉降剂,而且还作为抗结剂添加于食用盐中。 近年来,以K4[Fe(CN)6]为氰源和卤代芳烃反应制备芳基腈[11,12,13]和芳酰氯反应制备芳酰基腈[14]中已经被证明是一种很有效的氰化剂,参与反应时无需经过复杂的预处理,同时对反应液也无需进行特别处理,不会对环境造成污染。
本文报道一种以芳酰氯为原料,亚铁氰化钾为绿色氰化试剂,采用一锅两步反应合成一系列芳甲酸氰基芳甲酯的合成方法。 合成路线如Scheme 1所示。
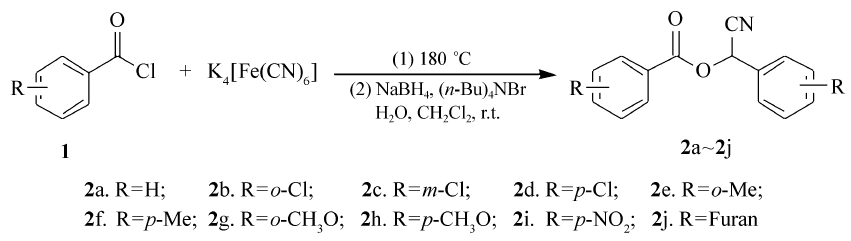 | Scheme 1 Synthesis of targart coumpounds |
K4[Fe(CN)6]·6H2O、苯甲酰氯、对甲基苯甲酰氯、邻甲基苯甲酰氯、间甲基苯甲酰氯、邻氯苯甲酰氯、间氯苯甲酰氯、对氯苯甲酰氯、呋喃酰氯、硼氢化钠、二氯甲烷、乙酸乙酯、石油醚、乙醇和甲醇均购自阿拉丁试剂有限公司,均为分析纯试剂;薄层色谱(TCL)使用的GF254 荧光硅胶板购自青岛海洋化工有限公司,试剂级;柱层析采用粒径为45~75 μm硅胶购自青岛海洋化工有限公司,化学纯。
Alpha Centauri FI-IR型红外光谱仪(美国Mattson仪器公司);Mercury-400 plus型核磁共振仪(NMR,德国BRUKER分析仪器公司);ZF-7A型精密熔点仪(上海光学仪器一厂);ZF-1型三用紫外分析仪(上海骥辉科学分析仪器有限公司)。
在装有回流冷凝管的50 mL圆底烧瓶中加入2.24 g(16 mmol)的苯甲酰氯与1.474 g(4 mmol)的K4[Fe(CN)6],油浴180 ℃反应3 h。 将混合物冷却至室温,加入10 mL二氯甲烷,搅拌并逐滴加入5 mL H2O与0.091 g(2.4 mmol)硼氢化钠和0.155 g(0.48 mmol)四丁基溴化铵的混合溶液。 加毕后室温搅拌6 h。抽滤,滤液浓缩,硅胶上柱,洗脱液为 V(石油醚): V(乙酸乙酯)=20:1,得目标化合物。
化合物2a 白色固体(Lit.[5] off-white solid);mp 56~58 ℃。 IR(KBr), σ/cm-1:3054,2959,2244,1727,1600,1495,1452,1269,1101,752,712;1H NMR(CDCl3,400 MHz), δ:6.68(s,1H,CH),7.45~7.50(m,6H,Ar),7.60~7.65(m,2H,Ar),8.06~8.09(m,2H,Ar);13C NMR(CDCl3,100 MHz), δ:63.3,116.2,127.8,128.0,128.6,129.2,130.0,130.4,131.8,134.1,164.6。
化合物2b 白色固体(Lit.[5] white solid);mp 50~52 ℃。 IR(KBr), σ/cm-1:3064,2969,2256,1740,1591,1476,1438,1311,1232,1097,1036,767,739;1H NMR(CDCl3,400 MHz), δ:6.96(s,1H,CH),7.33~7.51(m,6H,Ar),7.81~7.84(m,1H,Ar),7.91(d,J=7.5 Hz,1H,Ar);13C NMR(CDCl3,100 MHz), δ:61.1,115.1,126.8,127.4,127.7,129.2,129.7,130.3,131.5,131.9,132.0,133.5,133.8,134.7,163.1。
化合物2c 浅黄色液体(Lit.[5] oil)。 IR(oil), σ/cm-1:3058,2959,2247,1737,1601,1484,1376,1243,1089,1042,756,732;1H NMR(CDCl3,400 MHz), δ:6.63(s,1H,CH),7.41~7.51(m,4H,Ar),7.59~7.63(m,2H,Ar),7.94~7.98(m,1H,Ar),8.02~8.04(m,1H,Ar);13C NMR(CDCl3,100 MHz), δ:62.9,115.4,126.0,128.0,128.2,129.4,130.0,130.1,130.7,130.8,133.2,134.3,134.9,135.3,163.3。
化合物2d 浅黄色固体(Lit.[5] light yellow solid);mp 64~65 ℃。 IR(KBr), σ/cm-1:3076,2954,2236,1743,1598,1465,1369,1234,1096,798,773;1H NMR(CDCl3,400 MHz), δ:6.62(s,1H,CH),7.43~7.47(m,4H,Ar),7.54(d,J=8.4 Hz,2H,Ar),7.97(d,J=8.4 Hz,2H,Ar);13C NMR(CDCl3,100 MHz), δ:62.7,115.5,126.1,129.0,129.2,129.4,130.0,131.2,136.6,140.7,163.5。
化合物2e 白色固体;mp 50~52 ℃。 IR(KBr), σ/cm-1:3036,2938,2240,1726,1450,1232,1050,734;1H NMR(CDCl3,400 MHz), δ:6.33(s,1H,CH),2.39(s,J=8.4 Hz,3H,CH3),3.74(s,J=8.4 Hz,6H,CH3),6.99~7.86(m,8H,Ar);13C NMR(CDCl3,100 MHz), δ:13.9,14.1,63.6,114.9,124.7,125.4 ,127.7,128.4,128.9,129.1,129.6,130.8,131.2,132.7,138.2,138.9,167.0。
化合物2f 白色固体;mp 72~74 ℃。 IR(KBr), σ/cm-1:3248,2894,1694,1458,1250,1186,1034,738;1H NMR(CDCl3,400 MHz), δ:2.41(s,3H,CH3),2.43(s,3H,CH3),6.63(s,1H,CH),7.27(d,J=6.4 Hz,4H,Ar),7.51(d,J=8.4 Hz,2H,Ar),7.95(d,J=8.4 Hz,2H,Ar);13C NMR(CDCl3,100 MHz), δ:65.2,118.7,121.7,125.3,129.1,130.6,136.2,139.6,147.9,154.6,166.2。
化合物2g 淡黄色固体;mp 104~105 ℃。 IR(KBr), σ/cm-1:3358,2922,2228,1724,1600,1436,1232,1006,742;1H NMR(CDCl3,400 MHz), δ:6.25(s,1H,CH),3.72(s,J=8.4 Hz,3H,CH3),3.74(s,,J=8.4 Hz,6H,CH3),6.70~7.86(m,8H,Ar);13C NMR(CDCl3,100 MHz), δ:56.0,56.3,60.2,113.3,114.0,114.9,115.7,116.1,120.0,120.7,128.8,130.0,130.7,133.8,162.5,163.2,167。
化合物2h 白色固体;mp 64~66 ℃。 IR(KBr), σ/cm-1:3036,2920,2228,1904,1724,1612,1512,1240,1076,752;1H NMR(CDCl3,400 MHz), δ:6.25(s,1H,CH),3.73(s,J=8.4 Hz,3H,CH),6.70~7.86(m,8H,Ar);13C NMR(CDCl3,100 MHz), δ:56.0,70.1,113.3,114.9,122.4,122.8,130.7,161.3,166.3,167.0。
化合物2i 白色固体;mp 72~74 ℃。 IR(KBr), σ/cm-1:3039,2918,2247,1736,1601,1489,1345,1218,1122,774,734;1H NMR(CDCl3,400 MHz), δ:6.32(s,1H,CH),7.26(d,J=8.4 Hz,2H,Ar),8.14(d,J=8.4 Hz,2H,Ar),8.23~8.34(m,4H,Ar);13C NMR(CDCl3,100 MHz), δ:65.2,118.7,121.7,125.3,129.1,130.6,136.2,139.6,147.9,154.6,166.2。
化合物2j 黄色固体;mp 120~122 ℃(Lit.[5] 122~124 ℃)。 IR(KBr), σ/cm-1:3038,2923,2228,1732,1496,1354,1280,1098,782,743;1H NMR(CDCl3,400 MHz), δ:6.47~6.49(m,1H,Fu),6.54~6.56(m,1H,Fu),6.72(s,1H,CH),6.75~6.77(m,1H,Fu),7.33~7.34(m,1H,Fu),7.52~7.54(m,1H,Fu),7.65~7.67(m,1H,Fu);13C NMR(CDCl3,100 MHz), δ:55.7,111.1,112.2,113.0,116.8,120.6,142.4,145.1,146.3,147.7,156.2。
以苯甲酰氯为原料,K4[Fe(CN)6]为氰化试剂,硼氢化钠为还原剂的反应为模板,探讨了第2步反应的物料配比,反应温度和反应时间对产率的影响。 结果见表1。
表1 苯甲酸氰基苯甲酯的合成 Table 1 Synthesis of mandelonitrile benzoate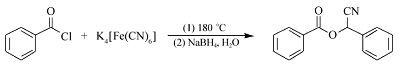 |
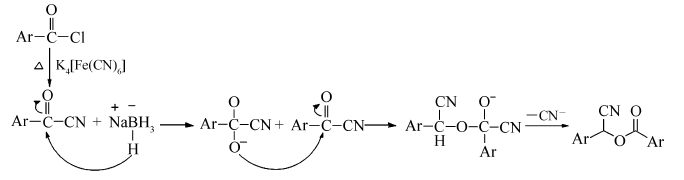
首先,在无溶剂条件下苯甲酰氯和亚铁氰化钾直接反应合成苯甲酰腈。 此步反应的温度必须高于150 ℃,否则反应难以发生,这主要是因为亚铁氰化钾中Fe3+和CN-之间键能较大,低温下很难将其键打断而释放氰基。 为了使反应时间缩短,反应更彻底,经多次实验,得出该反应的最佳温度为180 ℃,最佳反应时间为3 h,且只有当苯甲酰氯的用量为亚铁氰化钾用量的4倍化学计量时,产物的收率最高。 第2步反应中采用二氯甲烷、甲苯、四氢呋喃、乙酸乙酯、 N, N-二甲基甲酰胺(DMF)作为溶剂,其中在乙酸乙酯、乙醇中没有目标产物出现,而在四氢呋喃和 N, N-二甲基甲酰胺中,能得到目标产物,但产率不高。 通过几种溶剂的反应效果对比,发现二氯甲烷中作为溶剂时反应的效果最好。 另外,在该反应中对几种相转移催化剂PEG-400、四丁基氯化铵、四丁基溴化铵也进行了筛选,得出四丁基溴化铵在该反应中的效果最好。 第2步反应的温度对产率也有较大影响。 因为该反应是放热反应。如果升高温度,反应逆向进行,也会使产物的收率下降,所以,通过实验得出第2步反应在室温下进行6 h,产率最高达到72.5%(表1,Entry 8)。 通过探索,整个反应的合理条件为:第1步投料摩尔比 n(苯甲酰氯): n(亚铁氰化钾)=4:1,180 ℃反应2 h,第2步以CH2Cl2为溶剂,NaBH4为还原剂在四丁基溴化铵相转移催化下反应6 h。
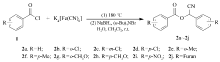
在上述反应条件下,对底物芳酰氯进行了扩展,不同芳酰氯和亚铁氰化钾经一锅两步反应,均得到了相应的芳甲酸氰基芳甲酯的目标产物(表2)。 反应中发现,芳酰氯苯环上的取代基对目标产物的收率影响较大,当苯环上带吸电子基时的收率高于供电子基时的收率,相同取代基在苯环的不同位置时,反应产率也有影响:对位>间位>邻位。 另外,杂环呋喃酰氯也能很好的发生反应生成呋喃甲酸氰基呋喃甲酯(表1,Entry 10)。
表2 芳甲酸氰基芳甲酯的合成 Table 2 Synthesis of aryl formic acid cyano aryl methyl esters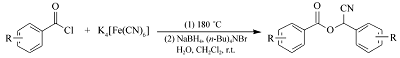 |
{kind=link}
{kind=link}
以芳酰氯为原料,亚铁氰化钾为氰化试剂,采用一锅两步反应合成了一系列芳甲酸氰基芳甲酯。 该反应的合理条件为:第1步投料摩尔比 n(苯甲酰氯): n(亚铁氰化钾)=4:1,180 ℃反应2 h,第2步以CH2Cl2为溶剂NaBH4为还原剂在四丁基溴化铵相转移催化下反应6 h,产物收率达61.7%~80.3%。 本合成方法具有操作简单,产率高,环境友好等优点,为工业化生产提供理论依据。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|