
LIU Jia, PAN Rongrong, ZHANG Erhuan, et al. Mechanistic Understanding of Plasmon-induced Hot Electron Injection for Photocatalytic and Photoelectrochemical Solar-to-Fuel Generation[J]. Chinese Journal of Applied Chemistry, 35(8): 890-901

由于金属纳米晶表面等离子共振产生的热电子效应,金属/半导体异质纳米晶的可控合成对于增强半导体光催化与光电催化性能具有显著的促进作用。 本综述阐述了热电子产生与驰豫的微观机制,探讨了影响热电子在金属-半导体异质纳米晶中界面传递效率的关键因素及异质界面调控合成的重要性,简要介绍了热电子注入效应在光催化与光电催化制备太阳能燃料研究中的应用进展,分析了目前存在的主要问题并对该领域未来的发展趋势进行了展望。

Hot electrons derived from the surface plasmon resonance of metallic nanocrystals have been demonstrated to play a promising role in promoting the efficiency of photocatalytic and photoelectrochemical solar-to-fuel generation. In this review, we try to describe the underlying mechanisms of the generation and relaxation process of hot electrons, give a discussion on the key factors that affect the efficiency of hot electron injection from metal to semiconductor, and provide an overview of the research progress on hot electron-mediated photocatalytic and photoelectrochemical water splitting. This review also outlines the critical limitations in current studies and sheds light on the possible future developments in this research field.






能源是支撑人类经济和社会发展的重要基石。 由于煤、石油、天然气等传统化石能源的日益枯竭及其大规模使用所引起的严重环境问题,发展可再生洁净能源已成为一项全球性的重大战略任务。 作为分布最为广泛,且唯一能够在总量上替代化石能源的可再生能源,太阳能的开发利用对于解决当前人类面临的能源与环境问题具有极大的潜力[1,2]。 自1972年日本Fujishima和Honda教授[3]发现TiO2电极能够在光的驱动下分解水产生H2气以来,模拟自然界的光合作用过程,探索将太阳能转化为化学能(氢气、甲烷、一氧化碳等)的有效途径一直是国际前沿研究的热点和难点。 过去几十年间,为了提高对光能的吸收与转化效率,研究人员主要致力于开发具有宽光谱响应、高载流子分离与利用效率的半导体基光催化材料。 近些年来,科研人员利用Au、Ag、Cu等具有表面等离子共振(surface plasmon resonance,简称SPR)特性的金属的光响应来驱动或促进半导体的光-化学转化过程,为开发高效光催化材料提供了崭新的研究思路[4,5,6,7,8]。
具有特定结构的等离子金属纳米晶可在很宽的太阳能光谱范围内(涵盖紫外、可见、近红外光)具有强烈的光吸收,由此在金属表面引起的电荷密度集体振荡——即表面等离基元(surface plasmon),能够将入射光的电磁能量限域在亚波长尺度的空间范围内,并产生强烈的光与物质相互作用。 借助丰富的化学合成手段,通过设计合成金属-半导体异质纳米结构材料将等离子金属与半导体在纳米尺度内有效耦合,将有机会充分利用等离子金属的SPR效应增强半导体对入射光的吸收,促进光生电子-空穴分离,从而大幅提高光催化材料的光能转化与利用效率。 这种等离子金属与半导体的协同耦合作用,实质上是将等离子金属收集到的光能转移到半导体中实现电荷分离,进而引发其它物理与化学过程,如光催化与光电催化反应等。 研究表明,等离子金属与半导体间的协同耦合作用主要通过3种途径进行:1)热电子(或热空穴)注入机理;2)基于局域电磁场增强的电荷分离机理;3)基于局域电磁场增强的共振能量转移机理。 在同一个金属-半导体异质结构体系中,这3种作用机理可能同时存在,共同促进光生载流子浓度的增加及半导体表面光-化学、光电-化学反应速率的提高[4,7,9,10]。
在本综述中,我们主要集中探讨表面等离子共振的热电子注入机理,通过分析热电子的产生与驰豫过程,揭示影响热电子注入效率的主要因素,并通过对代表性研究成果的介绍,简要评述热电子注入效应在光催化、光电催化制备太阳能燃料中的应用进展及未来发展前景。
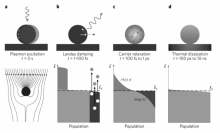
在初始平衡状态,金属纳米晶表面的电子服从体系温度下的费米-狄拉克分布。 具有特定波长的入射光电磁场可在金属表面诱发自由载流子的集体振荡,即产生局域表面等离子共振效应,使金属纳米晶能够从远大于其几何截面的空间范围内富集并且吸收光子(图1a)[4,7,11,12,13,14]。 光激发的等离子共振可通过两种途径衰减:1)辐射衰减:向外发射光子,即米氏散射(Mie scattering);2)非辐射衰减:一个等离子量子可通过朗道阻尼(Landau damping)在1~100 fs的时间尺度内激发一对热电子-热空穴对。 如图1b所示,等离子诱导产生的电磁场能够激发一部分导带电子从占据轨道跃迁到高于费米能级的非占据轨道,形成整体上偏离平衡态的处于高能量位置的电子态分布。 这些高能的热电子可在100 fs到1 ps的时间尺度内通过电子-电子散射作用快速将能量分散给周围的低能电子,使更多的电子被热化,而其本身的能量则显著降低,最终形成比体系温度更高的类费米-狄拉克分布(图1c)。 随着热电子能量与运动速度的降低,其与周围声子的散射作用逐渐增强,热电子可通过电子-声子相互作用将能量传递给金属表面的其它原子,引起晶格振动,在100 ps到1 ns的时间尺度内引起金属纳米晶的局部加热(图1d),最终整个体系恢复到标准的费米-狄拉克分布。 由上述的热电子产生与驰豫机理可见,热电子在产生后会迅速通过一系列的电子-电子、电子-声子相互作用驰豫为低能电子,其寿命极短,在fs量级。 而在空间尺度上,热电子在电子的平均自由程(3~10 nm)内即会完成高能电子到低能电子的衰变,即热电子仅能存在于金属纳米晶的表面区域[15,16]。
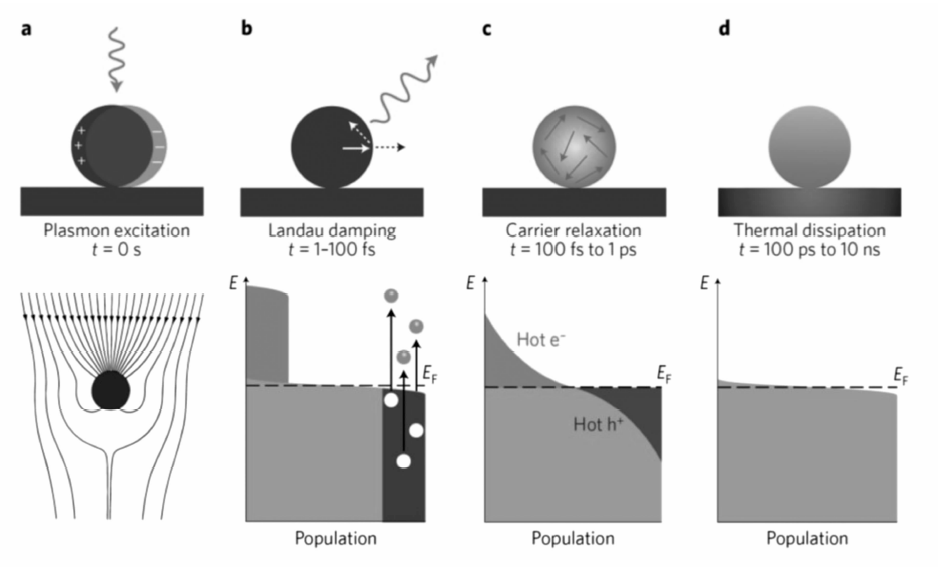 | 图1 金属纳米颗粒光致激发与驰豫过程的示意图及相应的特征时间尺度[4]Fig.1 Schematic illustration of the photoexcitation and subsequent relaxation processes following the illumination of a metal nanoparticle with a laser pulse and the characteristic timescales[4] (a)The excitation of a localized surface plasmon redirects the flow of light towards and into the nanoparticle. (b)In the first 1~100 fs following Landau damping, the athermal distribution of electron-hole pairs decays through carrier multiplication caused by electron-electron interaction. (c)The hot electrons will redistribute their energy by electron-electron scattering processes on a timescale ranging from 100 fs to 1 ps. (d)Finally, heat is transferred to the surroundings of the metallic structure on a longer timescale ranging from 100 ps to 10 ns via thermal conduction |
{kind=link}
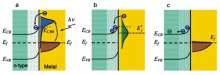
在金属与半导体(n型)形成的纳米异质结构中,金属的热电子注入半导体的机理与染料分子敏化相似,即等离子金属负责吸收光子,并将表面等离子共振效应产生的热电子传递至相邻半导体的导带[17,18,19,20,21,22,23]。 具体而言,热电子需要具有足够的能量,能够克服金属与半导体界面处形成的肖特基势垒(Schottky barrier)。 如图2a所示,在金属与半导体具有合适的能带排布的情况下,热电子在产生之初具有较高的能量,能够越过肖特基势垒注入相邻半导体的导带。 然而,由于电子-电子散射作用的发生,热电子能量迅速衰减,此时只有部分热电子仍然具有足够的能量克服肖特基势垒注入半导体(图2b)。 随着热电子能量进一步由于电子-电子、电子-声子散射作用衰减,整个体系恢复到平衡状态(图2c)。 从以上分析可见,热电子注入过程的时间非常短暂,仅能发生在表面等离子共振激发后的几百fs内,此外,热电子在金属与半导体界面间的高效传递需满足以下几个条件:1)金属与半导体间具有合适的能带排布,即热电子的初始能量能够克服界面肖特基势垒注入半导体导带;2)金属与半导体直接接触,且具有较大的接触面;3)金属与半导体形成的异质界面具有较少的晶格缺陷或杂质,能够有效抑制界面缺陷对热电子的捕获,实现热电子在界面间的快速传递。 2005年,日本的Tatsuma教授课题组[24]首次提出Au纳米颗粒等离子共振产生的激发电子能够转移至相邻半导体TiO2的导带,之后的十余年间,基于热电子注入效应的plasmon增强光催化与光电催化引起了广泛的研究兴趣,以下两节将针对这两个领域的研究进展分别做简要评述。
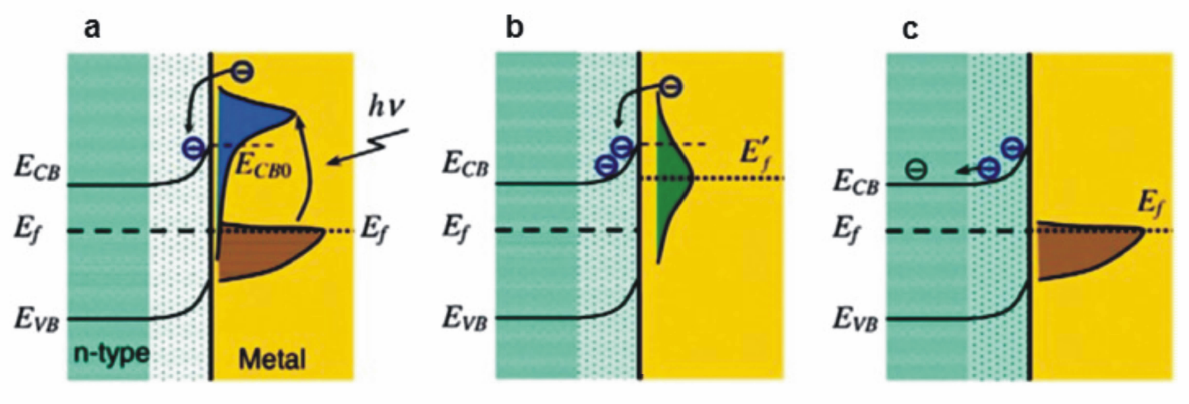 | 图2 热电子注入机理示意图[7]Fig.2 Schematic illustration of the hot electron injection effect[7] (a)Excitation of the electrons from thermal equilibrium to a high-energy state upon absorbing photons and injection of the electrons to the CB of the semiconductor. (b)Redistribution of electron energy and formation of a Fermi-Dirac distribution at a high-temperature Fermi level after the injection of the electrons. (c)Restoration of the standard electron distribution with electrons and holes flowing to different regions in the semiconductor |
{kind=link}
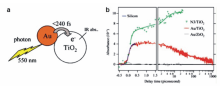
2007年,日本的Furube教授[25]利用飞秒泵浦瞬态吸收光谱观测到了plasmon激发的热电子从Au纳米颗粒跃迁到TiO2的直接证据。 如图3a所示,在550 nm的可见光激发下,直径为10 nm的Au纳米颗粒产生高能的热电子,能够越过Au与TiO2之间约1.0 eV的肖特基势垒,注入到TiO2的导带中。 根据TiO2导带电子对中红外光的特征吸收,可利用波长为3500 nm的中红外光作为探测光,检测在550 nm泵浦光激发的条件下,从Au跃迁到TiO2导带的热电子的数量。 如图3b所示,该体系在前240 fs出现了强烈的瞬态吸收信号,表示热电子从Au注入TiO2导带的过程发生在等离子共振激发的飞秒尺度范围内。 以敏化效率为100%的钌基配合物染料分子(简称N3)与TiO2形成的复合体系为参照,可估算热电子从Au注入TiO2导带的产率为40%。 此外,作者还研究了Au的热电子向ZrO2导带的转移过程,发现在同样的激发与探测条件下Au/ZrO2体系没有明显的瞬态吸收信号,说明Au的热电子无法有效注入ZrO2导带,可能是因为ZrO2具有较高的导带位置(比TiO2导带高0.9 eV),热电子的能量无法克服界面处的肖特基势垒注入ZrO2。
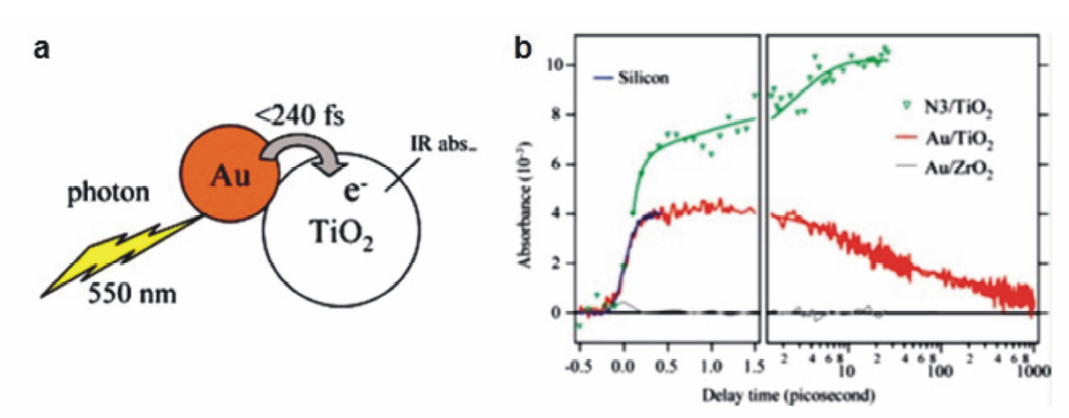 | 图3 (a)热电子从Au纳米颗粒注入相邻TiO2纳米颗粒的示意图。(b)不同纳米晶薄膜样品在3500纳米探测光下的瞬态吸收动力学图谱(绿色N3/TiO2,红色:Au/TiO2,灰色:Au/ZrO2)[25]Fig.3 (a)Schematic diagram of a gold nanodot attaching on a TiO2 nanoparticle, also indicating the revealed electron injection process. (b)Transient absorption kinetics at 3500 nm of nanocrystalline films(green:N3/TiO2, red:Au/TiO2, gray: Au/ZrO2)[25] |
{kind=link}
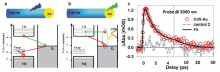
2013年,美国连天泉教授课题组[26]采用飞秒泵浦瞬态吸收光谱系统研究了Au纳米颗粒(5.4 nm)/CdS纳米棒(26.7 nm×3.6 nm)异质结构中等离基元与激子的相互作用机理。 他们首先利用波长为400 nm的泵浦光选择性激发异质结构中CdS纳米棒的带间跃迁,并采用波长为3000 nm的光源探测CdS中激发电子向Au的转移过程(图4a)。 另一方面,他们利用波长为590 nm的泵浦光选择性激发异质结构中Au的表面等离子共振,考察热电子从Au向CdS的跃迁动力学过程(图4b)。 实验结果表明,CdS中的激发电子向Au的转移速率具有较宽的动力学分布,整个过程可持续数个皮秒,且这种电荷分离的状态可保持长达1.5 μs。 相反,表面等离子共振激发产生的热电子从Au向CdS的迁移则是发生在97 fs内的超快动力学过程(图4c),并且热电子在注入CdS导带后寿命非常短暂,约1.8 ps后即会迁移回Au与空穴发生复合。 此外,他们定量计算了热电子从Au向CdS的注入效率[26]:其中Δ A(590 nm)与Δ A(400 nm)分别为590 nm泵浦光激发Au/CdS异质结构与400 nm泵浦光激发CdS纳米棒所产生的瞬态吸收信号大小;OD(590 nm)与OD(400 nm)分别为Au/CdS异质结构在590 nm及CdS纳米棒在400 nm的光密度; j(590 nm)与 j(400 nm)分别为590 nm与400 nm泵浦光的光子通量。 计算结果表明,在该体系中热电子从Au纳米颗粒注入CdS纳米棒的量子效率为2.75%,作者提出,进一步优化异质结构中Au与CdS各自的尺寸与形貌将是提高热电子注入效率的可行途径。
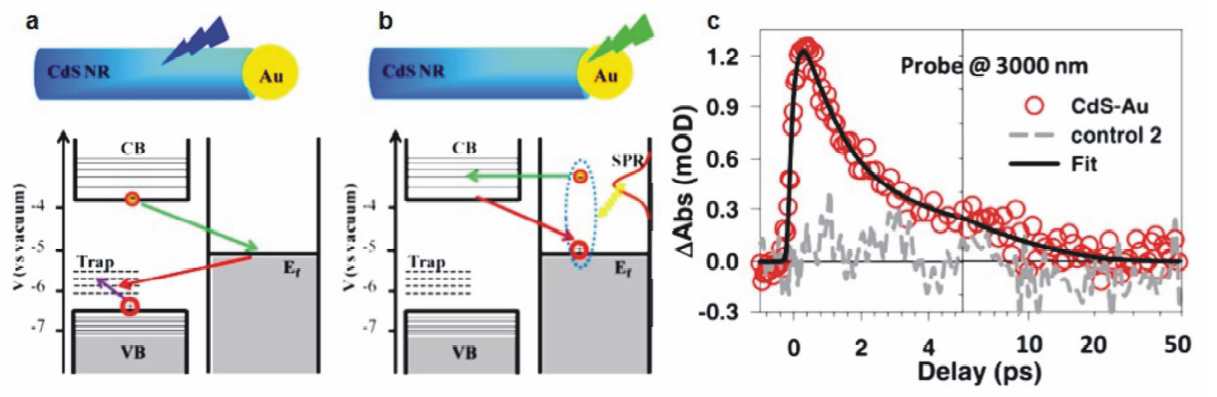 | 图4 (a)选择性激发Au/CdS异质结构中CdS 纳米棒的激子跃迁引起的电荷分离示意图。(b)选择性激发Au/CdS异质结构中Au纳米颗粒的等离子共振引起的电荷分离示意图。(c)Au/CdS纳米棒异质结构与对比样品2(CdS纳米棒与Au纳米颗粒的物理混合物)在3000 nm探测光下的瞬态吸收动力学图谱[26]Fig.4 (a)Charge separation induced by exciting the excitonic transition in the CdS nanorod domain. (b)Charge separation induced by plasmonic excitation in the Au tip. (c)Kinetics probed at 3000 nm for Au/CdS nanorods and control 2(a mixture of CdS nanorod and Au nanoparticle)[26] |
{kind=link}
我们课题组[27]针对金属/半导异质结构的可控合成开展了系列研究,在前期工作的基础上,我们近期成功发展出一种水相阳离子交换非外延生长方法,能够克服Au与CdS间高达42.7%的大晶格失配度,制备出一类在界面结构、壳层结晶性、结构可控性与传统晶种生长法所合成的核壳结构截然不同的Au@CdS核壳纳米晶。 如图5a~5f所示,所合成的Au@CdS纳米晶具有紧密接触、原子级有序排列的界面结构,同时CdS半导体壳层表现出罕见的准单晶性。 飞秒泵浦瞬态吸收光谱研究表明,这种原子级洁净的异质界面能够有效避免传统Au@CdS核壳结构中常见的界面缺陷与杂质对热电子的捕获,十分有利于促进热电子从Au向CdS的快速转移(图5g和5h)。 计算表明,该体系中热电子从Au核注入CdS壳层的效率高达48%,远高于已知其他文献报道中Au/CdS异质结构的热电子注入效率。 此外,这种新型合成方法能够实现核壳纳米晶中金属与半导体尺寸(Au核直径:5~35 nm连续可调;CdS壳层厚度:2~10 nm连续可调)与形貌(纳米球、纳米棒)的独立精确调控。 有限元模拟实验证实,这种对纳米晶结构的高度可控性为调节Au与CdS各自的电子结构与光学特性,增强等离基元-激子耦合提供了十分有效的手段。 基于以上优异特性,所合成的Au@CdS核壳纳米晶在可见光下表现出高效的光催化产氢性能,其活性比传统方法合成的Au@CdS核壳结构提高了2~3个数量级单位,说明该新型合成方法在plasmon增强太阳能转化与利用研究中具有广阔的应用前景。
 | 图5 (a~c)球形与(d~f)棒状Au@CdS核壳纳米晶的高分辨透射电子显微镜照片与高角度环形暗场扫描电子显微镜照片。图(c)与图(f)分别为图(b)与图(e)中黄色方框区域的放大图片。(g)热电子从Au等离基元注入CdS导带的示意图,以及中红外飞秒泵浦瞬态吸收光谱测试的原理图。(h)Au@CdS核壳纳米晶(Au核直径35 nm,CdS壳层厚度10 nm)在3900 nm探测光下的瞬态吸收动力学图谱[27]Fig.5 HRTEM and HAADF-STEM images of (a~c)spherical and (d~f)rod-like Au@CdS nanocrystals. (c) and (f) are magnified images corresponding to the areas enclosed by the yellow squares in (b) and (e), respectively. (g)Schematic illustration of the hot electron injection process from Au plasmon to CdS conduction band, and the principle of the Mid-IR femotosecond pump-probe transient absorption spectroscopy measurement. (h)Transient absorption kinetics probed at 3900 nm for Au@CdS nanocrystals with 35 nm-sized Au core and 10 nm-sized CdS shell[27] |
{kind=link}
除了等离子金属与TiO2、CdS等传统半导体的耦合,近日江海龙教授课题组[28]报道了类n型半导体多孔材料——金属有机框架材料(metal-organic framework,简称MOF)MIL-125与Au纳米棒及Pt纳米粒子共同形成的异质结构在光催化产氢中的优异性能。 首先,他们在合成MIL-125的过程中原位引入Pt纳米粒子得到Pt@MIL-125结构,然后利用静电吸引作用将所得材料与Au纳米棒集成得到Pt@MIL-125/Au结构。 如图6a所示,这种合成路线能够实现异质结构中Au纳米棒与Pt纳米粒子在空间上的有效分离。 紫外-可见漫反射实验表明,MIL-125具有与宽禁带半导体类似的光响应行为,其自身不能被可见光激发,但MIL-125与Au纳米棒及Pt纳米粒子耦合形成异质结构后,在可见光照射下表现出161.3 μmol/(g·h)的光催化分解水产氢活性与良好的稳定性(图6b),说明Au纳米棒的热电子能够有效转移至MIL-125的LUMO轨道用于光催化反应。他们进一步通过电子自旋共振技术考察了Au纳米棒与Pt纳米粒子对于增强MIL-125光催化性能的协同作用,研究结果表明,Pt纳米粒子与MIL-125形成的肖特基结能够定向引导热电子在MIL-125上的迁移,从而有效提高光生电子与空穴的空间分离(图6c),实现可见光下光催化效率的大幅提升。
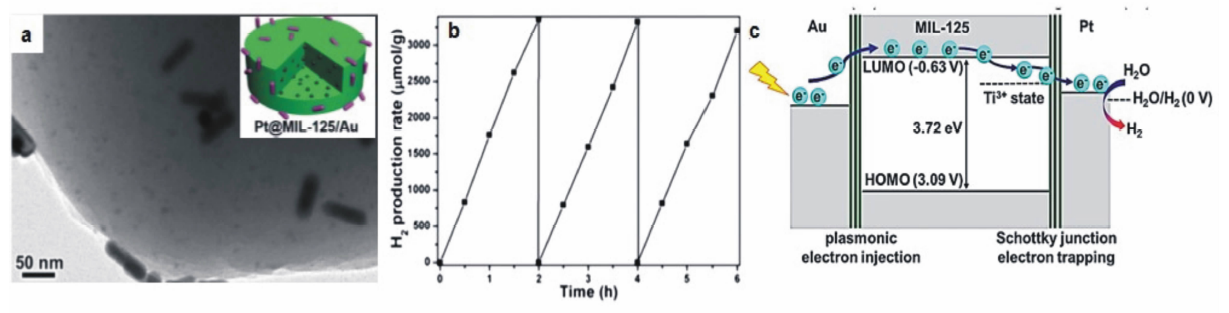 | 图6 (a) Pt@MIL-125/Au的透射电子显微镜照片。(b)Pt@MIL-125/Au在380~800 nm可见光激发下光催化产氢的活性与稳定性表征。(c)Pt@MIL-125/Au样品中光生电子在Au-MOF与Pt-MOF界面处的迁移示意图[28]Fig.6 (a)TEM image of Pt@MIL-125/Au. (b)Activity and recycling performance of Pt@MIL-125/Au in photocatalytic H2 production under 380~800 nm light irradiation. (c)Schematic illustration showing the electron migration at the two metal-MOF interfaces[28] |
{kind=link}
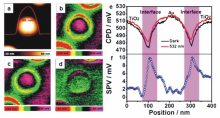
随着研究的深入,热电子参与n型半导体光还原半反应的机理已得到了充分的论证,然而,与热电子同时产生的热空穴在光氧化半反应中所扮演的角色则鲜有报道。 2017年,李灿院士课题组[29]利用先进的开尔文探针力显微镜技术(KPFM)结合光沉积实验与密度泛函理论(DFT)模拟,对Au/TiO2异质光催化材料中热空穴的空间分布与在光催化水氧化反应中的作用机理进行了系统探索。 他们发现,光沉积实验能够直观地区分出热电子与热空穴在Au/TiO2中的不同空间分布。 在光沉积实验中采用可见光选择性激发材料中Au的表面等离子共振,可以观察到易于捕获电子的Cr2O3主要沉积于TiO2表面,而易于捕获空穴的PbO2则主要沉积于Au与TiO2的界面以及Au的表面,说明Au等离子共振激发的热电子大多迁移至TiO2,而与热电子同时产生的热空穴则主要聚集于Au与TiO2形成的异质界面。 KPFM测试结果进一步证实了热空穴的界面分布(图7a~7d),将暗态下的KPFM图像与532 nm单色光激发下的KPFM的图像作差值,能够看出Au与TiO2形成的环形界面具有较高的表面电势(图7d),与表面光电压的测试结果相一致(图7e和7f),说明热空穴倾向于聚集在Au与TiO2的异质界面,削弱界面处的能带弯曲并引起功函数的下降。 根据DFT模拟计算的结果,该体系中在Au与TiO2界面处形成的Ti-O-Au特殊结构能够对热空穴起稳定作用,有效延长热空穴的寿命,同时Ti-O-Au可充当活性位,十分有利于促进对水分子的吸附与活化,提高光催化水氧化活性。
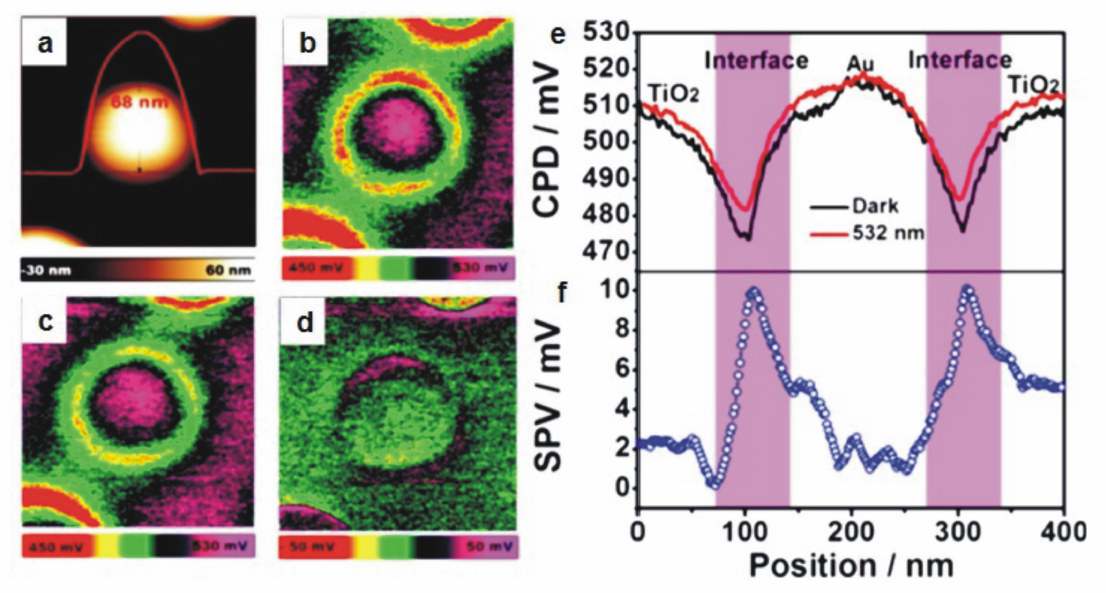 | 图7 (a)沉积在TiO2金红石上的Au纳米颗粒的原子力显微镜照片。Au/TiO2结构在(b)暗态与(c)532 nm单色光激发下的开尔文探针力显微镜照片。(d)将暗态下的图像与532 nm单色光激发下的图像作差值所得的表面光电压图像。(e)Au/TiO2结构在暗态(黑线)与532 nm单色光激发下(红线)的接触电势曲线。(f)将暗态下的曲线与532 nm单色光激发下的曲线作差值所得的表面光电压曲线[29]Fig.7 (a)Atomic force microscope topography image of Au nanoparticle deposited on a TiO2 rutile single crystal. KPFM images of Au/TiO2 (b) in the dark and (c) upon 532 nm illumination. (d)The surface photovoltage image by subtracting the potential under dark conditions from that under 532 nm illumination. (e)Contact potential difference profiles of dark(black line) state and light(red line) state across the Au/TiO2 particle. (f)The surface photovoltage profile across the Au/TiO2 particle by subtracting the potential under dark from that under 532 nm light irradiation[29] |
{kind=link}
与等离子金属与n型半导体形成的异质结构相比,由于肖特基结在界面处引起方向相反的能带弯曲,等离子金属与p型半导体形成的异质结构表现出不同的热载流子界面迁移行为。 熊宇杰教授课题组[30]以Ag与p型半导体BiOCl形成的异质结构为模型,研究了热载流子在该体系中的传输过程与光催化分解水产氧性能。 如图8a所示,他们合成了具有单晶结构的BiOCl方形纳米片,其上、下表面为(001)晶面,4个侧面则为(110)晶面。 由于界面电荷分离效应,光生空穴倾向于从(001)晶面向(110)晶面定向迁移。 基于这种特殊的性质,他们进一步在BiOCl的(001)晶面选择性沉积Ag纳米立方块,而在(110)晶面选择性沉积Pd纳米立方块,形成Ag-(001)BiOCl(110)-Pd异质结构。 光催化分解水产氧实验表明,这种精心设计的异质结构具有优异的光催化性能,其产氧活性与BiOCl, Ag-(001)BiOCl以及 BiOCl(110)-Pd相比具有明显提高。 根据以上实验结果并结合第一性原理计算模拟结果,他们提出以下机理(图8c):在全光谱光源的照射下,可见光激发Ag纳米立方块的表面等离子共振,所产生的深层热空穴能够越过肖特基势垒注入BiOCl(001)晶面的价带;与此同时,紫外光激发BiOCl的带间跃迁,在其价带产生大量的光生空穴。由于界面电荷分离效应,从Ag注入的热空穴以及BiOCl(001)晶面带间跃迁产生的空穴能够定向迁移至BiOCl(110)晶面的价带,与(110)晶面自身产生的空穴汇合;得益于BiOCl与Pd纳米立方块间形成的向下能带弯曲,这些富集在(110)晶面上的空穴能够被Pd纳米立方块捕获,有效降低光生电子-空穴复合的几率,促使Ag-(001)BiOCl(110)-Pd异质结构在光催化水氧化反应中表现出显著增强的产氧活性。
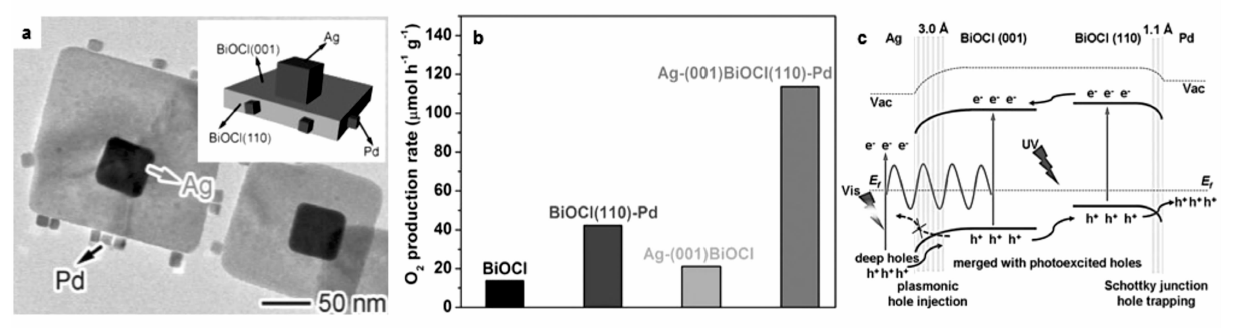 | 图8 (a)Ag-(001)BiOCl(110)-Pd异质结构的透射电子显微镜照片。(b)全光谱下BiOCl、BiOCl(110)-Pd、Ag-(001)BiOCl与Ag-(001)BiOCl(110)-Pd的光催化水氧化产氧活性。(c)Ag-(001)BiOCl(110)-Pd异质结构的能带排布示意图以及光生电子在Ag-(001)BiOCl 与BiOCl(110)-Pd异质界面的迁移示意图[30]Fig.8 (a)TEM image of Ag-(001)BiOCl(110)-Pd hybrid structure. (b)Photocatalytic O2 evolution from water with BiOCl, BiOCl(110)-Pd, Ag-(001)BiOCl and Ag-(001)BiOCl(110)-Pd as catalysts under full-spectrum irradiation. (c)Schematic illustrating the band alignments and charge flow at two metal-semiconductor interfaces[30] |
{kind=link}
热电子注入效应在增强光电催化的研究中同样引起了广泛的兴趣[31,32,33,34,35]。 2012年,美国Moskovits教授课题组[36]报道了TiO2与产氧助催化剂共同修饰的Au纳米棒阵列光阳极在光电催化分解水中的应用。 他们首先利用电沉积法在ITO/TiO2导电基底上生长垂直取向的Au纳米棒阵列,之后利用电子束沉积法在Au纳米棒的顶端生长TiO2薄层,并通过电化学沉积法在Au纳米棒的侧面担载钴基产氧助催化剂,所形成的Co-OEC/AuNR/TiO2光阳极的形貌与结构如图9a~9e所示。 他们发现,在紫外光照射下,所制备的光阳极仅产生0.02 mA/cm2的光电流,而在可见光照射下,光电流信号显著增加,可达到0.35 mA/cm2(图9f)。 此外,图9f显示Co-OEC/AuNR/TiO2光阳极在可见光下的光电流信号由快响应与慢响应两部分构成。 由于TiO2本身只具有紫外光响应活性,从以上结果可推测,Au纳米棒表面等离子共振激发产生的热电子能够越过肖特基势垒注入TiO2导电基底,并在外加偏压作用下迅速转移至对电极Pt网上发生水还原产氢反应,而留在Au纳米棒上的空穴则能够在钴基产氧助催化剂作用下迅速发生水氧化产氧反应,产生快响应光电流信号(图9g)。 与此同时,Au纳米棒产生的热电子也可以注入顶端沉积的TiO2薄层,这些被困在顶端的光生电子最终能够转移至对电极Pt网发生产氢反应,并在这一过程中形成慢光电流响应信号。 他们提出,经过以上两个过程,最终约95%的热电子能够被用于可见光下的水分解反应。
 | 图9 在ITO/TiO2导电基底上生长Co-OEC/AuNR/TiO2纳米棒阵列光阳极的(a)结构示意图,(b)扫描电子显微镜照片与(c~e)透射电子显微镜照片。(f)Co-OEC/AuNR/TiO2光阳极分别在紫外光与可见光激发下的光电流-时间曲线。(g)Co-OEC/AuNR/TiO2光阳极的能级结构示意图[36]Fig.9 (a)Schematic illustration, (b)SEM image and (c~e)TEM images of Co-OEC/AuNR/TiO2 nanorod arrays on ITO/TiO2 glass. (f)Photocurrent vs time plots for Co-OEC/AuNR/TiO2 photoanodes under UV and visible light irradiation, respectively. (g)Energy band diagram of Co-OEC/AuNR/TiO2 photoanode[36] |
{kind=link}
2014年,美国吴年强教授课题组[37]报道了具有三明治结构的CdS-Au-TiO2纳米棒阵列光阳极在光电催化分解水中的优异性能。 如图10a~10d所示,他们首先利用水热合成法在FTO基底上生长TiO2纳米棒阵列,再利用光还原法在TiO2纳米棒上生长尺寸为11.5 nm的Au纳米颗粒,最后利用化学浴沉积法在外层沉积CdS量子点薄层,形成CdS-Au-TiO2三明治结构纳米棒阵列光阳极。 研究发现,这种特殊结构的光阳极在模拟太阳光全光谱照射下能够产生4.07 mA/cm2的光电流,而同样条件下CdS-TiO2纳米棒阵列光阳极的光电流仅为3.10 mA/cm2。 此外,在275~375 nm紫外光激发下以及波长大于425 nm的可见光激发下,CdS-Au-TiO2纳米棒阵列光阳极均表现出比CdS-TiO2更高的光电流(图10e)。图10f给出了与CdS-TiO2纳米棒阵列光阳极相比,引入Au纳米颗粒形成三明治结构对光电转换效率(incident photoelectron conversion efficiency,简称IPCE)的增强曲线。 可以看出,在325~525 nm的波长范围内,IPCE的增强因子保持在1.5左右,而在525~725 nm范围内,IPCE增强因子显著上升,与Au纳米颗粒的SPR吸收峰相吻合。 他们进一步利用瞬态吸收光谱技术系统考察了CdS-Au-TiO2与CdS-TiO2两种体系中的不同电荷传输行为,提出Au纳米颗粒对于增强CdS-Au-TiO2三明治结构纳米棒阵列的光能转化效率起到了双重作用:1)在波长小于525 nm的光源激发下,CdS与TiO2同时产生带间跃迁,Au纳米颗粒可充当电子继电器,促进光生电子从CdS向TiO2的迁移(图10g);2)在波长为525~725 nm的光源激发下,Au纳米颗粒受激产生表面等离子共振,所产生的热电子能够克服界面肖特基势垒注入TiO2导带,从而将CdS-TiO2的光响应范围从525 nm拓宽至725 nm(图9h)。
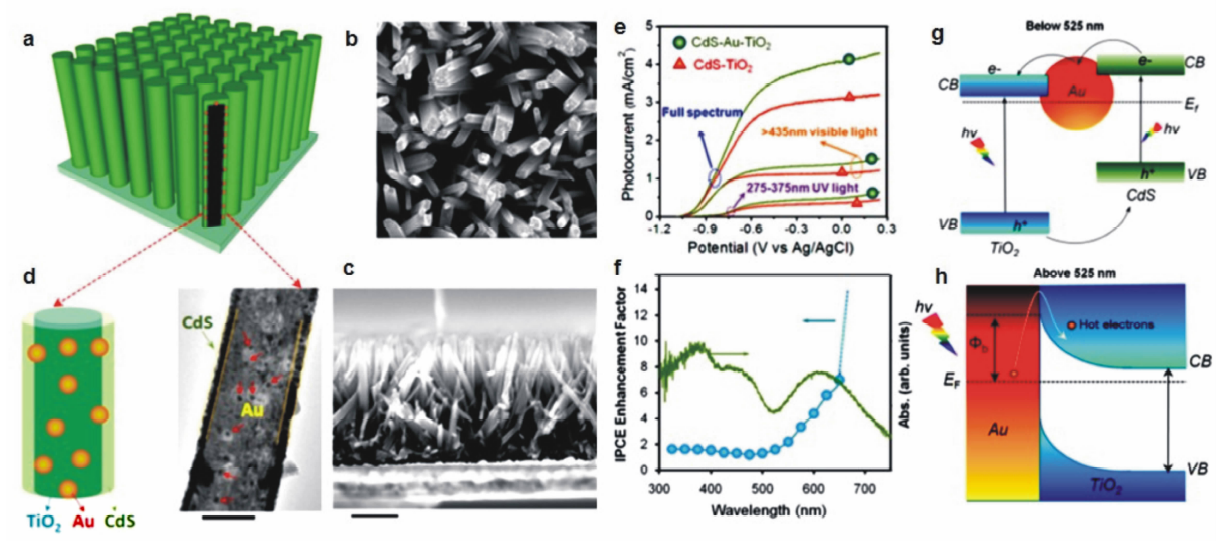 | 图10 CdS-Au-TiO2三明治结构纳米棒阵列光阳极的(a)结构示意图、(b)顶视图与(c)侧视图扫描电子显微镜照片。 (d)CdS-Au-TiO2三明治结构纳米棒的透射电子显微镜照片。 (e)CdS-Au-TiO2三明治结构纳米棒阵列光阳极分别在模拟太阳光全光谱,可见光(>430 nm)及紫外光(275~375 nm)照射下的光电流-偏压曲线。 (f)CdS-Au-TiO2三明治结构纳米棒阵列光阳极的IPCE增强曲线。 (g)三明治结构光阳极中Au纳米颗粒的电子继电效应示意图,在波长小于525 nm的光源照射下,Au纳米颗粒能够促进光生电子从CdS向TiO2转移。 (h)在波长大于525 nm的光源照射下,表面等离子共振产生的热电子能够从Au纳米颗粒注入TiO2[37]Fig.10 (a)Scheme for the CdS-Au-TiO2 sandwich nanorod array on the FTO substrate. (b)Top-view and (c)cross-section view of the CdS-Au-TiO2 nanorod array. (d)TEM image of a single sandwich nanorod. (e)Photocurrent-applied potential (J-V) curves for CdS-Au-TiO2 nanorod array irradiated by full-spectrum of simulated solar light, visible light(>430 nm) and ultraviolet light(275~375 nm), respectively. (f)IPCE enhancement for CdS-Au-TiO2 nanorod array. (g)Electron relay effect of Au nanoparticles, facilitating the charge transfer from CdS to TiO2 nanorod under the irradiation of incident solar light with wavelength <525 nm. (h)Plasmonic energy transfer from the excited Au nanoparticle to TiO2 through the hot electrons transfer under the irradiation of incident light with wavelength >525 nm[37] |
{kind=link}
新加坡陈晓东教授与李述周教授共同合作[38],合成了具有多级分段结构的CdS-Au纳米棒阵列光阳极用于光电催化分解水研究。 他们采用阳极氧化铝为硬模板,通过电化学方法在ITO基底上交替生长CdS-Au-CdS-Au-CdS-组分,形成具有多级分段结构的纳米棒光阳极(图11a和11b)。 以离散偶极近似的模拟结果为指导,他们将所合成的分段结构光阳极中每段Au与CdS组分的长度分别控制在200 和400 nm,以实现有效的等离基元-激子耦合。 采用形貌相近的纯CdS纳米棒阵列作为对比,他们发现具有CdS-Au多级分段结构的纳米棒阵列在模拟太阳光全光谱照射下能够产生更高的光电流,且光电流的大小与分段的数目成线性增加的关系(图11c),最终光电流可高达10.5 mA/cm2。 IPCE的测试结果则表明,多级分段结构光阳极的光电流增强与其中Au组分的表面等离子共振密切相关(图11d)。 由于CdS的导带与价带分别为-4.1与-6.5 eV,且CdS的费米能级为-4.3 eV,Au的费米能级为-5.1 eV,因此多级分段结构中CdS与Au的界面形成高度为0.8 eV的肖特基势垒(图11e)。 根据以上实验结果以及对能带结构的分析,他们提出了CdS-Au多级分段结构光阳极中的光生电荷转移模型(图11f和11g),并指出Au组分等离子共振引起的有效光生电荷分离是其表现出优异的光电转换性能的重要原因。
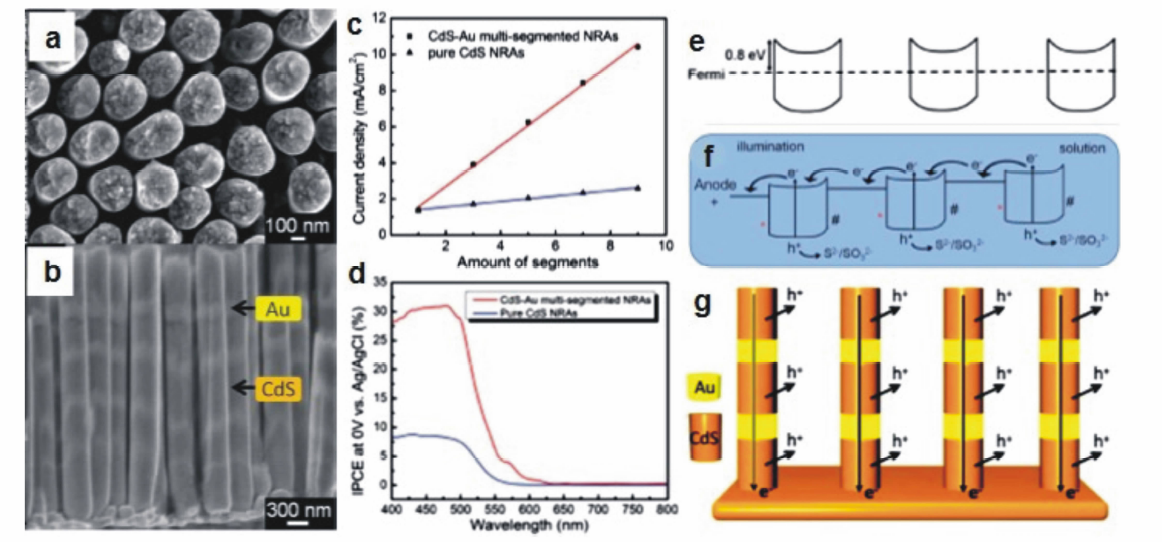 | 图11 CdS-Au多级分段结构纳米棒阵列光阳极的(a)顶视图与(b)侧视图扫描电子显微镜照片;(c)CdS-Au多级分段结构纳米棒阵列光阳极以及CdS纳米棒阵列光阳极的光电流与分段数目之间的线性关系;(d)在0 V vs Ag/AgCl的外加偏压下CdS-Au多级分段结构纳米棒阵列光阳极与CdS纳米棒阵列光阳极的IPCE曲线;(e) CdS-Au多级分段结构纳米棒中的费米能级排布示意图;(f,g)CdS-Au多级分段结构纳米棒阵列的光生电子传递路径示意图[38]Fig.11 SEM images of (a)the top view and (b)the side view of multi-segmented CdS-Au nanorod arrays photoanode. (c)The linear relationship between the photocurrent intensities and the amount of segments in the multi-segmented CdS-Au nanorod arrays photoanode and the pure CdS nanorod arrays photoanode. (d)Measured IPCE spectra of the multi-segmented CdS-Au and pure CdS nanorod arrays photoanodes at a potential of 0 V vs Ag/AgCl. (e)Schematic illustration of the Fermi level alignment in the multi-segmented CdS-Au nanorod. (f,g)Schematic illustration of the photo-generated electron transfer process in each multi-segmented CdS-Au nanorod[38] |
{kind=link}
为了解决人类社会日益增长的能源需求与日益严峻的环境问题,如何通过光催化与光电催化的方式将太阳能高效转化为太阳能燃料,这一圣杯式的研究课题受到了各国科学家的高度关注。 近10年以来,密集的研究进展已经充分展示了金属的表面等离子共振效应对于增强半导体光催化与光电催化性能的重要作用与该领域广阔的研究前景。 然而,由于热电子在产生后会迅速驰豫耗散能量,其飞秒量级的寿命使得热电子注入半导体并最终参与表面化学反应的效率总体上仍处在较低的水平。 要提高热电子在太阳能燃料生产中的利用效率,需要研究者们对热电子产生与驰豫机制背后的物理原理有更深层次的认识,此外,要求研究者们对影响热电子注入效率的各项决定性因素开展更为系统深入的研究。 在以上原理的指导下,纳米合成技术的进一步发展也是提高热电子注入效率的关键一环,例如,如何更加精准可控的构建金属与半导体的纳米几何结构从而精准可控的调节其电子与光学特性,如何从原子层次设计构建金属与半导体之间的异质界面结构以促进热电子的快速界面迁移,如何科学的构建多重界面势垒以更加有效的引导热电子的定向迁移,如何通过设计合成多元化合物半导体以更加灵活的调控半导体电子结构及其与金属的耦合效率等等。 基于金属表面等离子共振在太阳能燃料生产中的巨大应用潜力,热电子注入效应在未来的几年仍将是一项研究热点,具有不同学科背景的研究者的协同合作将可能在这一领域带来科学技术上的重要突破。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|