

Haifeng TANG, Fengchao CUI, Lunyang LIU, et al. Insight into the Inhibitory Activities of Diverse Ligands for Tyrosinase Using Ligand- and Structure-based Approaches[J]. Chinese Journal of Applied Chemistry, 35(7): 788-794


酪氨酸酶是细胞内催化合成黑素的关键酶。 理解酪氨酸酶抑制剂结构与活性之间的关系对于设计新药和化妆品具有重要意义。 然而,酪氨酸酶抑制剂的定量构效关系仍不清楚。 本文利用配体和结构描述符构建了隐式和显式模型,阐明了酪氨酸酶抑制剂定量构效关系。 隐式模型的相关系数 R高达0.961,显式模型的相关系数为0.775。 两个模型很好地预测了3个茶多酚的酪氨酸酶抑制活性,表儿茶素没食子酸酯(ECG)>表没食子儿茶素没食子酸酯(EGCG)>没食子酸(G)。 相关性分析发现,抑制剂与酪氨酸酶结合引起的构象熵损失与抑制剂的活性密切相关。 具有较少构象熵损失的ECG在4种茶多酚中具有较高酪氨酸酶抑制活性。 结合自由能计算也证实ECG与酪氨酸酶的结合能力最强。 此外,通过分解结合自由能发现,酪氨酸酶活性中心的氨基酸残基(His57、His201、Asn202、His205、Glu192和Val215)与抑制剂形成了较强的范德华和静电相互作用,进而稳定了复合物结构。
The use of variant inhibitors to regulate the bioactivities of tyrosinase, which is the key enzyme in charge of the production of melanin and pigments, is a long-standing approach to design cosmetic and pharmaceutical products. The quantitative description of the structure-activity relationship of tyrosinase inhibitors is still unclear. In this study, we constructed descriptive models by integrating ligand- and structure-based approaches for such purpose. They provide correlation coefficients of 0.961 for implicit models and 0.775 for explicit model, respectively, to descript the activities of three tea polyphenols with the tyrosinase inhibitory activity order of (-)-Epicatechin gallate(ECG)>(-)-Epigallocatechin gallate(EGCG)>Gallic acid(G). As revealing from the descriptive models, entropy loss is more important than other features for determining inhibitory activity and thus the tyrosinase-ECG complex with the fewer conformational entropy loss has the strongest inhibitory activity in vitro among the four tea polyphenols. Moreover, residues including His57, His201, Asn202, His205 Glu192 and Val215 are the core of active sites in tyrosinase, and stabilize the tyrosinase-inhibitor complex by van der Waals and hydrogen bonding interactions.


酪氨酸酶(EC 1.14.18.1)广泛存在于动植物细胞和真菌中[1],其可以催化酪氨酸生成多巴色素[2,3]。 酪氨酸酶的催化活性与人体皮肤色素沉积、削皮后的果蔬褐变以及农作物害虫的成熟等生理过程密切相关[4]。 利用抑制剂调控酪氨酸酶的生物活性可以阻止这些不利的生理过程[5,6]。 因此,筛选高效、安全的酪氨酸酶抑制剂对于人体健康、果蔬贮运和农业生产具有重要意义。
在过去20年里,基于配体或者结构的方法常常被用于酪氨酸酶抑制剂的筛选,如定量构效关系模型(QSAR)[7,8] 、药效团模型[9]和分子对接[10,11]等。 QSAR等线性回归模型多是基于配体结构而构建的,构建过程中没有考虑蛋白质与配体的相互作用,因此在稳定性方面具有一定的局限性。 分子对接的方法能够提供抑制剂与蛋白质相互作用的结构信息,进而可以更好地理解抑制剂结构与活性的关系[12,13,14]。 但是,大多数的分子对接程序,诸如FlexX、DOCK和GOLD等,并不能可靠地预测活性中心含有金属离子的蛋白质与抑制剂的结合强度[15],因而单独用分子对接方法预测酪氨酸酶抑制剂活性并不能得到可靠的结果。 此外,多数QSAR模型均采用半数抑制浓度(IC50)作为响应变量,然而在不同的实验条件下测得的同一种抑制剂的IC50值有很大差异。 例如,Kolbe等[16]报道熊果苷和曲酸的IC50值分别为6500和500 μmol/L,而Curto等[17]报道的值分别为547和43 μmol/L。 因此,在构建QSAR模型过程中,使用IC50值作为响应变量时,应该对其进行归一化处理。
为了消除在不同实验条件下测抑制剂活性引起的系统误差,我们首先定义了一个新的变量RIC[RIC=lg (抑制剂的IC50值/阳性对照曲酸的IC50值)]来表示抑制剂的活性,并将RIC值作为构建QSAR模型中的响应变量。 然后,利用Discovery Studio 2.5计算了6个基于配体结构特征的描述符,同时利用分子重叠、能量最小化和自由能计算获得了5个能量项,作为基于结构的描述符。 最后,利用随机森林算法[18,19]和蒙特卡罗算法[20]分别构建了隐式和显式的QSAR模型,并预测了儿茶素(C)、表儿茶素没食子酸酯(ECG)、表没食子儿茶素没食子酸酯(EGCG)和没食子酸(G)对酪氨酸酶的抑制活性。
蘑菇酪氨酸酶、多巴( L-DOPA)、曲酸(kojic acid)、儿茶素、没食子酸、表儿茶素没食子酸酯和表没食子儿茶素没食子酸酯购自美国Sigma-Aldrich公司;磷酸二氢钾和氢氧化钾(北京化工厂);以上试剂均为分析纯。
Epoch型紫外-可见分光光度计(UV-Vis,美国BioTek仪器有限公司);LC-10ATVP型高效液相色谱仪(HPLC,日本Shimadzu仪器有限公司)。
1.2.1 酪氨酸酶抑制剂数据库的建立
我们对1980年以来所报道的酪氨酸酶抑制剂进行了筛选并建立数据库。 筛选原则如下:1)测量酪氨酸酶抑制剂的活性时使用蘑菇来源的酪氨酸酶和多巴作为底物;2)在相同的实验条件下同时测量了抑制剂和阳性对照曲酸的IC50值;3)抑制剂的抑制类型为竞争性抑制。 经过以上条件的筛选最终得到了46个抑制剂及其IC50值(见辅助材料表S1)。 抑制剂的初始三维结构由网页服务器Molinspiration Galaxy 3D Structure Generator v2013.02 beta(www.molinspiration.com)构建,然后利用量子化学计算软件Gaussin 09的B3LYP模块进一步优化[21],优化时采用6-31G(d,p)基组。 将优化后的分子结构输入Discovery Studio 2.5软件并计算了6个基于配体的描述符。
1.2.2 酪氨酸酶-抑制剂结构的生成及自由能计算
本实验使用的酪氨酸酶分子结构(PDB ID:4P6R[22])从PDB数据库中下载[23]。 该结构中含该酶的天然底物酪氨酸和2个Zn离子。 在模拟研究中用2个Cu离子取代了2个Zn离子。 为了得到酶与其另一个天然底物多巴的复合物,我们将多巴分子重叠到酶活性中心的酪氨酸上。 重叠时采取羟基匹配的方式。 根据羟基的方向,多巴与酪氨酸共有4种匹配方式(见辅助材料图S1)。 由于第2种匹配方式(见辅助材料图S1B)形成的复合物具有最低的结合自由能,因此选取该复合物作为与抑制剂匹配的模板。 随后将46个经过结构优化的抑制剂分子分别重叠到酶活性中心的多巴分子上。 重叠过程中仍然采取羟基匹配方式,每个抑制剂分子与多巴匹配时会产生多种匹配结构。 利用NAMD (v 2.9)软件将经过分子重叠产生的酪氨酸酶-抑制剂复合物利用共轭梯度下降法进行5000步的能量最小化[24]。 然后利用分子力学-Poisson Boltzmann表面积法(MM-PBSA)对优化后的复合物结构进行自由能计算[25]。 复合物总的自由能变化可以由式(1)进行计算:
式中,分子力学能量(Δ EMM)包括范德华作用(Δ EvdW)、静电作用(Δ Eele)和键能项(Δ Eint)[26]。 Δ Eint项是键、角和二面角能量的总和,其在单轨迹计算中被抵消,可以忽略。 溶剂化作用项(Δ Gsol)包括极性溶剂项(Δ Gpolar)和非极性溶剂项(Δ Gnonpolar)。 Δ Gpolar由泊松-玻尔兹曼方程进行估算[27]。 其中,立方体格间距设为0.05 nm,外部介电常数设为80.0,溶质介电常数设为4.0。 Δ Gnonpolar由溶剂可及表面积线性方程式(2)进行拟合:
式中, γ和 b分别设置为0.5 kcal/(mol·nm2)和0.86 kcal/mol。 构象熵损失项(- TΔ S)利用AmberTools13中的Normal模块进行估算[26]。 更加详细的实验参数设置可以参考我们的前期工作[28]。 由于每个抑制剂分子与多巴能够生成多种匹配结构,选取一个总自由能最低的复合物作为最优结构进行分析。
1.2.3 隐式模型和显示模型的建立
将6个基于配体的描述符和5个基于结构的能量项合并组成向量组并作为自变量(表1),利用RIC作为响应变量分别构建隐式模型和显式模型。 构建隐式模型时采用随机森林算法的回归模式[18,19],构建显式模型时采用蒙特卡罗算法[20]。 为了验证模型的预测效果,我们选取了C、ECG、EGCG和G进行活性测试并对模型进行外部验证。 对于ECG、EGCG和G的活性按照已经报道的光谱法进行测量[29,30]。 在测量C的抑制活性时,反应体系有较深的颜色背景难以在光吸收中进行扣除,因此利用高效液相色谱对其进行活性测试。 以上测试均使用多巴作为酪氨酸酶的底物。
| 表1 原子或基团的描述符(属性) Table 1 Descriptors(features) of atoms or group employed in this work |
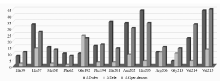
研究发现,所有抑制剂分子均结合在酪氨酸酶活性中心的铜离子附近。图1展示了代表性的酪氨酸酶与抑制剂(I25)形成的复合物结构。 酪氨酸酶活性中心的氨基酸残基主要通过范德华力和静电作用稳定抑制剂分子。 为了比较酶活性中心氨基酸残基对形成酪氨酸酶-抑制剂复合物时的能量贡献,我们对复合物总自由能进行分解并对具有较大的范德华或静电作用的氨基酸残基进行统计分析(Δ Evdw 或Δ Eele≥1.0 kcal/mol)。 如图2所示,在多数酪氨酸酶-抑制剂复合物中,有6个关键氨基酸残基可以与抑制剂产生较强的范德华力或静电力。 其中,Glu192主要通过静电引力与抑制剂相互作用。 其它氨基酸残基(His57、His201、Asn202、His205和Val215)则以范德华力为主。
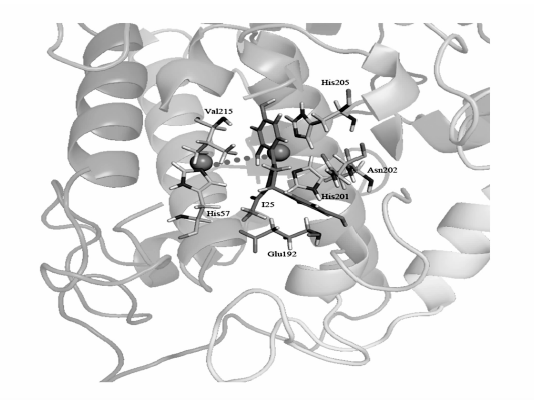 | 图1 典型的酪氨酸酶-抑制剂复合物结构Fig.1 Representative tyrosinase-inhibitor complex structure The tyrosinase-inhibitor complex was stabled by three types of interactions, i.e. the van der waals interaction between I25 and His57、His201、Asn202、His205、Val215, the hydrogen bond between I25 and Glu192, and the electronic force between the hydroxyl group on I25 and bi-copper ions inside the active center of tyrosinase |
{kind=link}
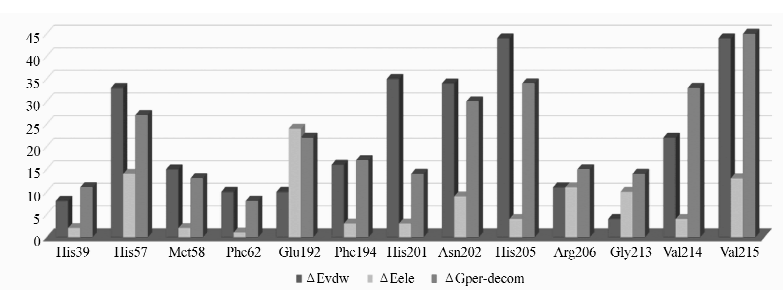 | 图2 能够形成强的残基-抑制剂作用(Δ Evdw 或Δ Eele >1.0 kcal/mol)的复合物数量对比分析(以范德华(Δ Evdw)、静电作用(Δ Eele)和总的自由能(Δ Gper-decomp)为例)Fig.2 The number of inhibitors stabilized by the residues with significant energy contributions no less than 1.0 kcal/mol for residue-inhibitor binding free energy(Δ Gper-decomp), van der Waals interaction(Δ Evdw), and electrostatic interaction(Δ Eele), respectively |
{kind=link}
本文所选用的描述符(自变量)与RIC(响应变量)之间的相关性具有较大差异,相关性如图3所示。 相关系数的绝对值越大说明结构属性与抑制活性之间的相关性越高,特征向量中包含的与抑制活性有关的信息越多。 在11个基于配体和结构的描述符中,构象熵(- TΔ S)、氢键受体数量(C_HBA)和羰基氧的电子态值(SES_dO)与抑制剂活性具有较高的正相关性。 这表明调控以下几个因素有利于提高抑制剂的活性:1)降低复合物形成时的构象熵损失;2)增加抑制剂分子中氢键受体数目;3)设计分支较多或长链线性抑制剂分子以提高羰基氧电子态值。 其中,构象熵项对提高抑制剂活性影响更为显著。
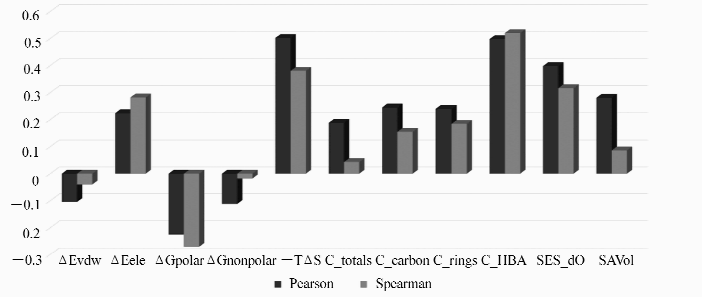 | 图3 基于配体与结的构属性与RIC之间的Pearson相关系数和Spearman相关系数Fig.3 The Pearson and Spearman correlation coefficients of ligand- and structure-based features against RIC |
{kind=link}
利用基于配体和结构的描述符、利用RIC值作为响应变量构建了隐式和显式模型,并将两个模型的预测值与实验值进行线性拟合。隐式模型得到了较高的相关性性系数( R=0.961),而显式模型的相关系数( R)为0.775(图4)。 由此可见,结构特征与抑制活性之间并不能用简单的线性关系加以描述。
 | 图4 隐式模型和显式模型的预测值与实验值之间的线性拟合曲线Fig.4 The correlation plots fitted values against experimental RIC values in the implicit model using Random Forest algorithm and the explicit model by Monte Carlo method |
{kind=link}
为了评估两个模型的预测能力,我们测量了天然产物C、ECG、EGCG和G的IC50值(如表2所示)。 活性测试时选取了来源于蘑菇的酪氨酸酶并以多巴为底物[32,33]。表2中也列出了利用隐式和显式模型预测的4个分子的RIC值。 除了C之外,预测的ECG、EGCG和G的酪氨酸酶抑制活性顺序与实验结果相一致。 ECG具有最高的酪氨酸酶抑制活性。 从辅助材料表S2中可以发现,相比于G,ECG和EGCG具有较高的羰基氧电子态值和氢键受体数,并且形成复合物时构象熵损失较少,这表明ECG和EGCG的酪氨酸酶抑制活性高于G,其实验和预测结果相一致。 同时发现,ECG和EGCG的羰基氧电子态值和氢键受体数大致相等,但ECG在形成复合物时有较少的构象熵损失,这进一步证明了形成复合物时较少的构象熵损失有利于提高抑制活性。 化合物C的预测值与实验值之间出现了较大偏差。这可能是由于C在活性测量过程中被消耗所导致的。 C是酪氨酸酶的一类底物[34,35],在抑制酪氨酸酶催化多巴时自身也被消耗,因此其IC50实验值可能会被高估。 值得一提的是,在这4个天然产物中,ECG具有最高的酪氨酸酶抑制活性,更具应用前景。
| 表2 天然产物及阳性对照的IC50值、RIC及其预测值 Table 2 RIC value of nature compounds and positive control from testing in vitro and predicting by models |
本文定义了相对抑制浓度(RIC)对抑制剂绝对活性IC50值进行归一化,以消除不同实验体系引入的系统误差。 同时本文利用分子重叠和能量最小化形成的蛋白-抑制剂复合物计算结合自由能使计算准确性和计算时间成本进行折中,实现了大量复合物自由能计算。 同时,将能量项与配体结构属性描述符合并形成的特征向量作为自变量,以RIC为响应变量,利用随机森林和蒙特卡罗算法分别构建了隐式模型和显式模型。 通过比较特征向量和抑制活性(RIC)之间的相关性发现复合物的构象熵损失对抑制活性具有重要影响。 当抑制剂与酪氨酸酶形成的复合物具有较小的构象熵损失时,可以产生较强的抑制作用。 另外,酚羟基等功能基团可以与酪氨酸酶活性中心的Cu离子配位占据活性中心,从而阻止天然底物进入活性中心。 同时,我们还发现酶活性中心的氨基酸残基His57、His201、Asn202、His205、Glu192和Val215能够通过与抑制剂间的范德华和氢键作用来稳定酪氨酸酶-抑制剂复合物。 通过对ECG、EGCG和G的预测和结构分析,上述结论被进一步证实。
辅助材料(Supporting Information)[酪氨酸酶抑制剂数据库和4种茶多酚的结构属性数据]可以免费从本刊网站(
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|