

DENG Huiyun, WANG Bin, WU Mao, et al. Preparation and Selective Recognition Property of Magnetic Surface Molecularly Imprinted Polymers with Gallic Acid as Template[J]. Chinese Journal of Applied Chemistry, 35(5): 600-608


羟基苯甲酸类化合物用途广泛,极性较强,在复杂水溶液体系中这些类似物的分离纯化与分析非常困难。 本文以磁性Fe3O4纳米颗粒为载体,没食子酸(GA)为模板分子,制备了磁性表面分子印迹聚合物(MIP)。 利用透射电子显微镜、红外光谱、磁强测定等检测手段对MIP进行了结构表征。 并对其吸附性能进行研究,比较了该MIP对GA及其它3种结构类似物的吸附性能差异。 结果表明,制备的以GA为模板的磁性分子印迹聚合物为核壳球形结构,键合牢固,对GA的吸附动力学符合准二级动力学方程模型,吸附过程属于Langmuir单分子层吸附。 该聚合物对GA表现出优异的选择性识别能力,其吸附量(318 K时37.736 mg/g)远远高于结构类似物。 该磁性分子印迹聚合物对模板分子不仅具有特异识别能力,而且能够磁控分离,分离效率高,可用于固相萃取。
Hydroxybenzoic acid compounds are used widely and have strong polarity. The separation, purification and analysis of their analogues in complex aqueous solution are very difficult. In this paper, magnetic surface molecularly imprinted polymer(MIP) was prepared using nano magnetic Fe3O4 as the carrier and gallic acid(GA) as the template molecule, and characterized by transmission electron microscopy, infrared spectroscopy and magnetic intensity measurement. Then its adsorption properties and adsorptive selectivity were studied by the static adsorption and dynamic adsorption experiments to compare the adsorption properties of GA on MIP with those of 2,4-dihydroxybenzoic acid, salicylic acid and benzoic acid. The results show that the MIP with GA as the template has a core-shell structure with a strong bonding effect, the adsorption process belongs to Langmuir monolayer adsorption and the adsorption kinetics can satisfy the pseudo-second-order kinetic equation model. The MIP exhibits an excellent selectivity for GA, and its adsorption capacity(37.736 mg/g at 318 K) is much higher than those of other structural analogues. The MIP prepared by this method can not only recognize the template molecule, but also be magnetically controlled. The high separation efficiency is applicable to the solid phase extraction.

羟基苯甲酸类化合物是重要的化工原料,广泛应用于医药、食品、农药和染料等行业[1,2],主要包括:没食子酸(GA)、间羟基苯甲酸(3-HBA)、尼泊金酸(4-HBA)、2,4-二羟基苯甲酸(BRA)、原儿茶酸(PCA)和水杨酸(SA)等。 其中,GA是一种高活性的生物多酚物质[3],广泛应用于食品、制药、化工及轻工等行业[4,5,6,7]。 在我国,常采用水解单宁酸的方法制备GA,其传统工艺主要有:酸水解法、碱水解法、酶法和发酵法[8,9],分离纯化技术包括:蒸馏法、冷冻澄清法、有机溶剂萃取法和离子交换法[10],通过对以上技术的多元组合,可以得到纯度较高的GA。 但是,仍然不能满足复杂体系中痕量类似物的分离纯化,而且分析这些类似物往往有一定的难度。
分子印迹技术(MIT)是近年来迅速发展的一种分子特异性识别技术[11],目前分子印迹聚合物的制备技术分为包埋法[12]和表面分子印迹法[13,14]两大类。 其中包埋法主要包含:本体聚合、悬浮聚合、乳液聚合和沉淀聚合等方法[15,16,17],但是随着分子印迹技术研究的逐渐深入,人们意识到,通过包埋法制备的分子印迹聚合物存在颗粒大、印迹点位于内部和结合位点少等缺点。 表面分子印迹法可以分为:牺牲载体法、聚合加膜法和化学接枝法[18],通过该法合成的分子印迹聚合物由于比表面积大,具有更多的有效印迹点,且其印迹层比包埋法制备的更薄,解吸、吸附速率更快,并可消除模板泄露。 由于以上优势,表面分子印迹技术受到日益重视,在药物分离、传感器、固相萃取和食品安全检测等方面[19,20,21]具有广阔的应用前景。
本文采用表面分子印迹法,以纳米级磁性Fe3O4为载体,以GA为模板分子,4-乙烯基吡啶为功能单体,乙二醇二甲基丙烯酸酯为交联剂制备了磁性表面分子印迹聚合物(MIP,即Fe3O4@mSiO2/MIP),进行了结构表征并深入探讨了该磁性表面分子印迹聚合物对GA的选择识别能力。
TecnaiG2 F20型透射电子显微镜(TEM,美国FEI公司),振动样品磁强计(VSM,美国量子公司Squid VSM型);Nexus 870型傅里叶变换红外光谱仪(FT-IR,青岛科诺达仪器有限公司);L600型高效液相色谱仪(HPLC,北京普析通用仪器有限责任公司)。
乙二醇二甲基丙烯酸酯(EGDMA)、3-丙基甲基丙烯酸酯(MPS)、4-乙烯基吡啶(4-VP)、纳米级磁性Fe3O4均为上海麦克林生化科技有限公司生产。
甲醇、乙腈、溴化十六烷基三甲基铵(CTAB)均购自太仓沪试试剂有限公司。 正硅酸四乙酯(TEOS,西陇化工股份有限公司生产)、GA(北京百灵威科技有限公司生产)、乙醇(天津市恒兴化学试剂制造有限公司生产)、乙酸(广东光华化学厂有限公司生产)、丙酮(国药集团化学试剂有限公司)、偶氮异丁腈(AIBN,江苏强盛功能化学股份有限公司)。 2,4-二羟基苯甲酸、水杨酸、苯甲酸等均为分析纯试剂。
将50 mg纳米Fe3O4颗粒和500 mg CTAB分散于100 mL去离子水中,超声混匀30 min,然后在该溶液中缓慢加入1.0 mmol/L NaOH溶液450 mL,超声5 min后,于60 ℃机械搅拌30 min。 然后于液面下缓慢注入TEOS-乙醇(体积比为1:4)溶液2.5 mL,继续搅拌5 min。 室温下静置12 h,磁控分离收集Fe3O4@CTAB/SiO2聚合物。 将收集的Fe3O4@CTAB/SiO2聚合物分散于80 mL丙酮中,于80 ℃回流24 h,重复2次,且每次回流后须用去离子水反复清洗,最后50 ℃真空干燥8 h即得Fe3O4@mSiO2聚合物。 将干燥的Fe3O4@mSiO2聚合物置于MPS溶液(150 μL的MPS溶于40 mL 10%的乙酸溶液)中,于50 ℃机械搅拌5 h,磁控分离收集,用去离子水反复清洗,50 ℃真空干燥即得双键改性的Fe3O4@mSiO2聚合物。 在N2气吹扫保护下,将GA(0.25 mmol)与功能单体4-VP(1 mmol)溶于无水乙腈进行预组装,在4 ℃下静置12 h,备用。 然后,将双键改性的Fe3O4@mSiO2聚合物(50 mg),交联剂EGDMA(5 mmol)和引发剂AIBN(20 mg)分散于致孔剂无水乙腈(15 mL)中,冰上操作,N2气吹扫下将预组装溶液滴入上述溶液中,然后于60 ℃机械搅拌24 h。 聚合完成后,磁控收集Fe3O4@mSiO2/MIP聚合物,用无水乙腈反复清洗至上清液透明,然后用甲醇-乙酸(体积比9:1)洗脱,直至完全脱除模板分子。 最后,用甲醇洗至中性,50 ℃真空干燥,即得MIP。 以相同步骤制备以SA为模板的MIP。
在不添加模板分子的情况下,重复以上实验步骤制备对照聚合物(NIP)。
采用FT-IR分析实验各阶段聚合物表面的接枝键合情况,通过TEM观测分子印迹聚合物结构与尺寸变化,通过VSM测定来评价分子印迹聚合物制备前后磁强变化。
分别准确称取0.2000 g GA等4种标准品于100 mL容量瓶中,去离子水定容得标准储备液。 标准储备液用去离子水稀释得到分别为0.10、0.20、0.30、0.40、0.50、1.00和2.00 g/L的标准溶液,采用HPLC法检测。 以浓度为横坐标,峰面积为纵坐标,绘制标准曲线。
HPLC条件:色谱柱为Pgrandsil-STC-C18(4.6 mm×250 mm,50 μm), V(流动相为甲醇): V(0.1%磷酸水溶液)=6.94,流速1.0 mL/min,检测波长273 nm,柱温25 ℃,进样量20 μL。
1.5.1 吸附热力学实验 配制一系列浓度的GA水溶液(0.10、0.20、0.30、0.40、0.50、1.00、2.00和2.50 g/L),分别向3.00 mL的GA溶液中加入15 mg的MIP或NIP。 在不同温度(298、308和318 K)下恒温振荡3 h,待其充分吸附后,将吸附后的溶液磁控分离,取上清液过滤,再通过HPLC测定其峰面积,对照GA标准曲线,求出吸附后的溶液中GA浓度,平行测定3次。 GA的吸附量由式(1)计算。
式中, Qe(mg/g)为聚合物对目标物质的吸附量, ρ0(g/L)和 ρ(g/L)分别是目标物质的始末质量浓度, V(mL)为目标物质水溶液的体积, m(g)为分子印迹聚合物的质量。
1.5.2 吸附动力学实验 在30.00 mL的GA溶液(2.00 g/L)中加入150 mg干燥后的MIP聚合物,置于318 K下恒温振荡,于不同时间段磁控分离取样,HPLC测定其峰面积,对照GA标准曲线,求出吸附后的溶液中GA浓度,平行测定3次。 GA的吸附量由式(1)计算。
1.5.3 选择吸附性实验 分别往3.00 mL的GA、BRA、SA、苯甲酸(BA)溶液(浓度均为2.00 g/L)中加入15 mg MIP。 同时置于318 K下恒温振荡3 h,待其充分吸附后,固液磁控分离,取上清进行HPLC分析。 计算该聚合物对4种物质的平衡吸附量,平行测定3次。 重复以上步骤,测定以SA为模板的印迹聚合物对这4种物质的吸附量。平衡吸附量由式(1)计算;采用印迹因子[22]( α)评价聚合物的识别特异性, α的值由式(2)计算;用分离因子[22]( β)衡量识别的选择性, β的值由式(3)计算。
式中, QMIP和 QNIP分别为印迹和非印迹聚合物对GA 的吸附量(mg/g)。
式中, Qtemplate和 Qnon-template分别为印迹纳米粒子对模板分子及干扰物的吸附量(mg/g)。
另外,取3.00 mL的GA、BRA、SA与BA混合标准溶液(质量浓度均为2.00 g/L),向混合溶液中加入15 mg MIP,置于318 K下恒温振荡3 h,用HPLC检测混合液中各组分吸附前后的浓度变化,评价MIP对这4种结构类似物的选择性吸附能力差异。
1.5.4 MIP对黑茶茶汤中GA的吸附 称取1.0000 g干燥黑茶茶叶,于90 ℃蒸馏水浸泡20 min,离心分离,得黑茶茶汤。 取3.00 mL黑茶茶汤加入35 mg的MIP,298 K震荡3 h,用HPLC检测吸附前后黑茶茶汤中GA的浓度变化。
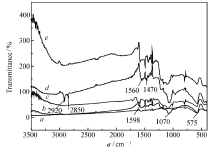
2.1.1 红外光谱分析 采用红外光谱分析了聚合物制备各阶段纳米颗粒的接枝键合情况。 由图1可知,所有纳米颗粒在575 cm-1附近均有吸收峰,为Fe3O4中Fe—O的特征吸收峰,说明后续聚合物成功包覆Fe3O4纳米颗粒。 将图1谱线 d与图1谱线 b红外光谱加以对比可以发现,Fe3O4@CTAB/SiO2(图1谱线 d)聚合物在2920和2850 cm-1处比Fe3O4@mSiO2(图1谱线 b)聚合物多出两个明显的吸收峰,为CTAB的特征吸收峰,说明结合在Fe3O4表面的CTAB经过丙酮回流,已经洗脱完全。 从图1谱线 b可以看出,在1070 cm-1左右的吸收峰为Si—O—Si反对称伸缩振动峰,表明二氧化硅层成功包裹在Fe3O4表面,而且CTAB也已经洗脱完全,从而推测在Fe3O4@mSiO2聚合物表面形成的二氧化硅壳层为多孔结构。
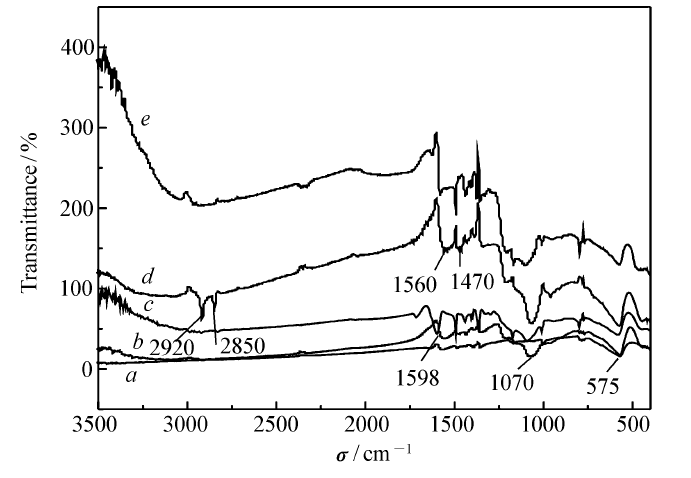 | 图1 分子印迹聚合物制备各阶段颗粒的红外光谱图 a.Fe3O4 nanoparticle; b.Fe3O4@mSiO2; c.Vinyl-modified Fe3O4@mSiO2; d.Fe3O4@CTAB/SiO2; e.MIPFig.1 Infrared spectra of surface molecularly imprinted polymers under producing process |
2.1.2 透射电子显微镜分析 Fe3O4纳米颗粒、Fe3O4@mSiO2和MIP的透射电镜如图2所示,可以清楚地看到聚合物的结构特点。 经TEM观测Fe3O4粒径约为200 nm,通过粒径分析仪检测粒径在180~220 nm的约占89%,粒径分布均匀。 从图2 B可以看出,Fe3O4@mSiO2颗粒为核壳结构,Fe3O4表面包裹的半透明状SiO2层约20 nm,且能明显看出均匀密布的孔道垂直分布于Fe3O4纳米颗粒的表面,与文献[23]报道中的硅系介孔材料一致,说明成功制备了介孔尺寸的Fe3O4@mSiO2;由图2 C知,MIP接近于球形,而且核壳结构明显,且修饰与印迹层厚度增大,粒径约为300 nm,后通过粒径分析仪检测粒径在270~330 nm的约占85%,粒径分布较均匀。 由此,通过聚合反应形成的聚合物已成功接枝到核壳结构外表面的SiO2层上,这说明已经成功制得核壳型磁性表面分子印迹聚合物。
 | 图2 透射电子显微镜照片 A.Fe3O4; B.Fe3O4@mSiO2; C.MIPFig.2 The images of TEM |
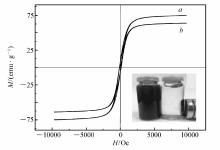
2.1.3 样品磁强测定图3为Fe3O4纳米颗粒和MIP的磁滞回线。 由图3可以看出,两条曲线的形态相似,没有剩磁,且矫顽磁力为零,说明二者均具有超顺磁性。 室温下,Fe3O4纳米颗粒和MIP的饱和磁化强度分别为74.7和63.6 emu/g,之所以MIP的饱和磁化强度比Fe3O4纳米颗粒的低,原因是其外表面包覆着非磁性外壳,对其磁响应产生了一定的影响。图3右下角的插图是MIP的磁控分离示意图,在没有外加磁场的情况下,MIP悬浮于水中,沉降速度缓慢;如果增加一个恒定磁场,MIP聚合物立即(15 s内)聚集到瓶壁,说明虽然MIP的磁性比Fe3O4纳米颗粒有所降低,但是对聚合物的快速磁控分离影响不大。
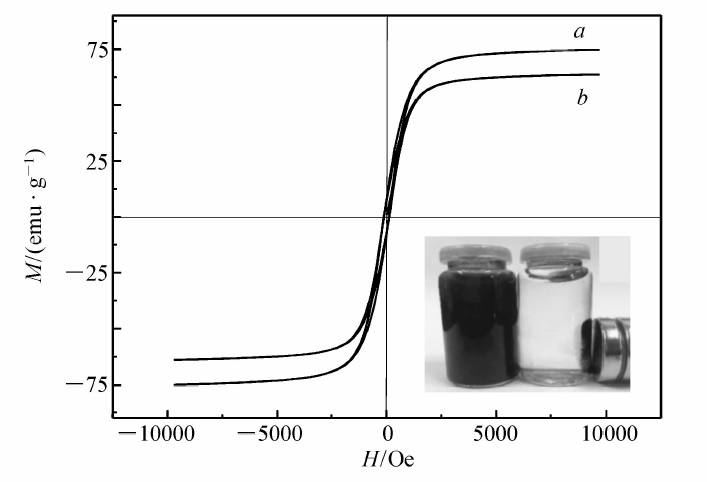 | 图3 Fe3O4纳米颗粒与MIP的磁滞回线图 a.Fe3O4 nanoparticle; b.MIPFig.3 The magnetization curves of Fe3O4 nanoparticle and MIP. Inset:the magnetic separation of MIP under the external magnetic field |
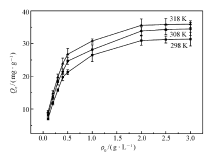
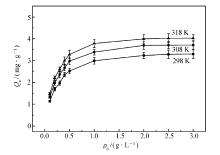
MIP和NIP在不同温度(298、308 和318 K)下的静态吸附曲线如图4和图5所示。 由图中可以看出,其吸附量随温度的升高而增加,原因可能是分子印迹聚合物在较高温度下聚合,在高温环境里有更好的吸附能力,与相关文献[24]报道的一致。 很明显,同一温度下,MIP的平衡吸附量比NIP的要大10倍以上。
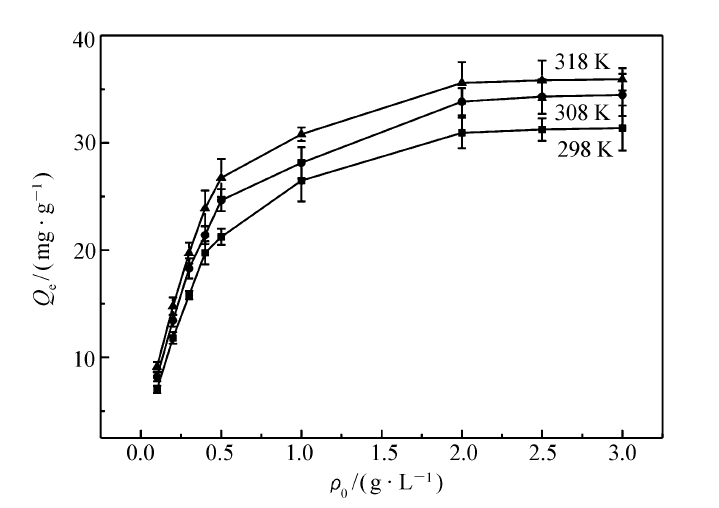 | 图4 MIP的静态吸附曲线Fig.4 The static adsorption curves of MIP |
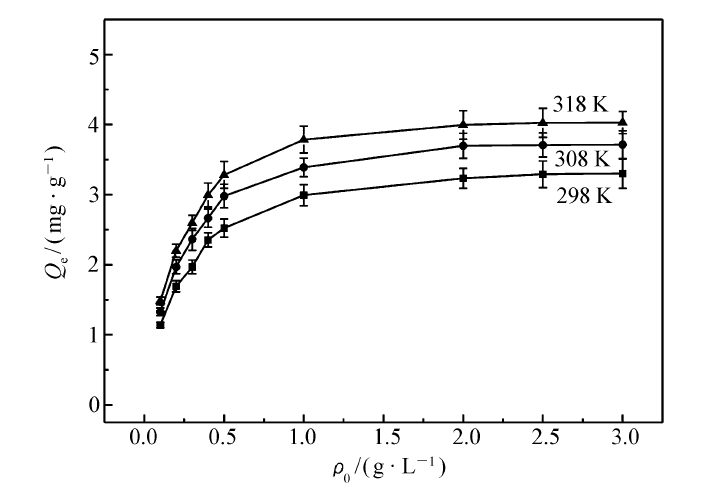 | 图5 NIP的静态吸附曲线Fig.5 The static adsorption curves of NIP |
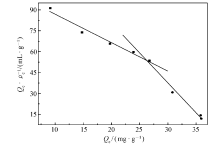
分别对MIP和NIP在不同温度下对不同浓度的GA水溶液的静态吸附数据进行了Langmuir方程(4)和Freundlich方程(5)拟合,拟合结果如表1所示。 并用Scatchard模型(6)对318 K时的MIP静态吸附进行拟合,分析结果如图6。
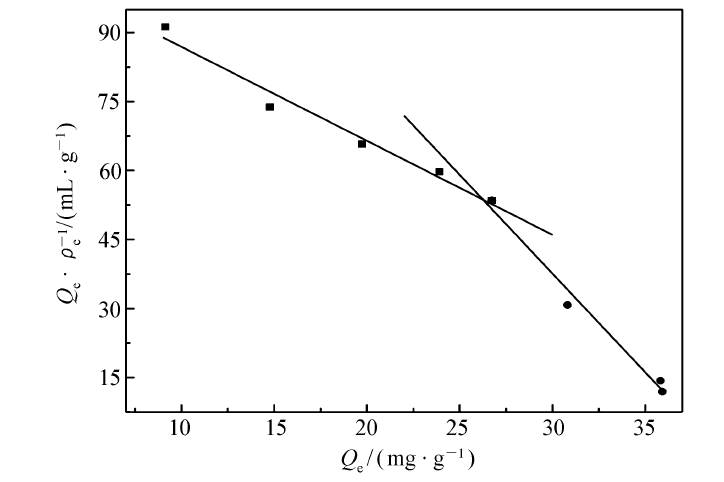 | 图6 Scatchard模型对MIP静态吸附分析Fig.6 Static adsorption analysis of MIP by Scatchard model |
Langmuir吸附等温方程:
Freundlich吸附等温方程:
Scatchard吸附等温方程:
式中, Qe、 Qm分别为吸附平衡时GA的平衡吸附量(mg/g)、饱和吸附量(mg/g), ρe为吸附平衡时GA的质量浓度(g/L), KL是Langmuir等温方程参数,代表表示吸附能力的强弱,其大小与吸附剂、吸附质的本性及温度高低有关; Kf是Freundlich吸附平衡常数,表示吸附剂一定时,吸附质为单位质量浓度时的吸附量; n为Freundlich特征吸附参数, KD为离解常数。
由Langmuir和Freundlich方程拟合的参数如表1所示。 Langmuir方程被用于描述吸附质在吸附剂表面的单分子层吸附过程,Freundlich方程引入了吸附质分子在吸附过程中的相互作用,常用来描述多层吸附过程。 由表1可知,相比于Freundlich吸附等温方程,Langmuir吸附等温方程具有更高的相关系数( R2>0.99),且通过Langmuir方程计算的饱和吸附量 Qm(cal)与实验所测得的饱和吸附量 Qe(exp)更接近。 证明该聚合物对GA的吸附过程是一个单分子层吸附过程。 由Scatchard拟合曲线(图6)可以看出, Qe/ ρe与 Qe呈非线性相关性,但可将图中的散点分成两部分,分别进行线性回归,得到两条直线,表明MIP中主要存在两类不同的结合位点。 根据直线的斜率和截距,分别计算出两类结合位点的平衡离解常数 KD和最大吸附量 Qmax。 对于高亲和力结合位点: KD1=0.4893 g/L, Qmax1=52.517 mg/g;对于低亲和力结合位点: KD2=0.2331 g/L, Qmax2=38.771 mg/g。
| 表1 Langmuir方程和Freundlich方程的参数 Table 1 Parameters of Langmuir equation and Freundlich equation |
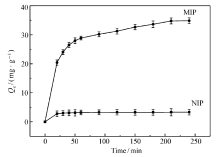
MIP在318 K下的吸附动力学曲线如图7所示。 由图7可知,MIP对GA的吸附量随吸附时间的延长而增加,特别是在60 min前急剧上升,但由于吸附过程需要缓慢进入孔道与识别位点结合,所以最终在180 min左右趋于平衡;而NIP的吸附过程为非特异性吸附,所以吸附量比较小,在50 min左右就已经趋于平衡。
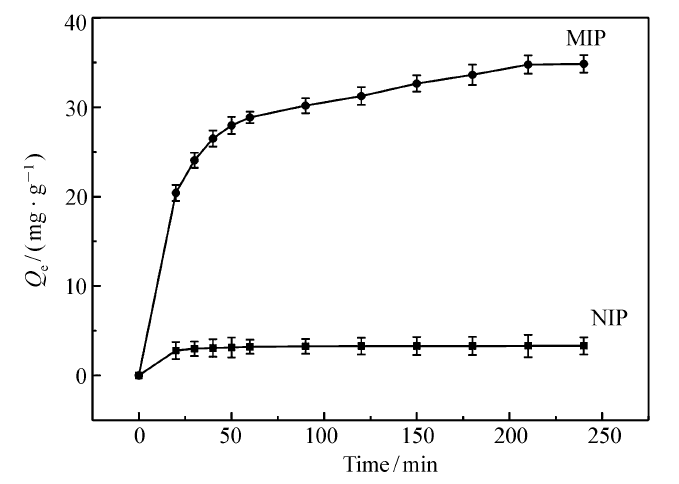 | 图7 吸附动力学曲线 MIP:Fe3O4@mSiO2/MIP, NIP:Fe3O4@mSiO2/NIPFig.7 The adsorption kinetics curves of MIP and NIP |
为了进一步研究MIP对GA的吸附动力学,将在318 K下的吸附数据用准一级方程和准二级方程进行拟合,得到表2的相关参数。 从表2可以看出,准二级动力学方程模型具有更高的相关系数( R2>0.99),且由准二级动力学方程计算的吸附量与实验所测得的吸附量很接近,可知MIP对GA的吸附行为符合准二级动力学方程,说明该吸附行为在一定浓度范围内吸附速率不仅与吸附质浓度正相关,而且与吸附剂MIP的量的大小正相关。 在吸附剂的量一定时,由于吸附剂表面的吸附位点数量有限,其吸附速率的增长受到限制,存在一个最大吸附速率。
| 表2 准一级、准二级动力学方程模拟参数(318 K) Table 2 Simulation parameters of pseudo first-order and pseudo two-order kinetic equations(318 K) |
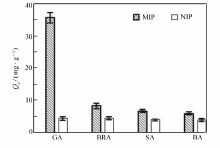
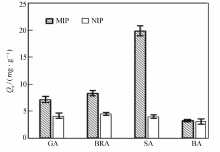
将分别以GA、SA为模板制得的MIP和NIP对GA、BRA、SA、BA的选择性吸附情况绘如图8和图9所示。 由图8和9可见,以GA为模板的MIP对GA表现出优异的选择性吸附作用,吸附量达35.58 mg/g,而对BRA、SA、BA的吸附量大大降低,分别为8.23、6.62和5.87 mg/g。 同样地,以SA为模板的MIP则对SA表现出优异选择性,对其它3种羟基苯甲酸类的吸附量很小。另外,NIP对各物质的吸附量无明显差别,说明MIP对模板分子具有较高的选择性吸附能力。 以GA为模板的MIP,GA初始浓度为2.00 mg/mL时,GA的其印迹因子 α=8.12,分离因子 βGA/BRA=4.32, βGA/SA=5.37, βGA/BA=6.06。 取吸附前后GA、BRA、SA与BA混合标准溶液,经HPLC分析,对比发现GA浓度明显减少,而其它3种物质浓度基本没有变化,进一步表明制得的磁性分子印迹聚合物对GA(模板分子)存在特异吸附选择性,可以很好地从结构类似物中识别出GA。
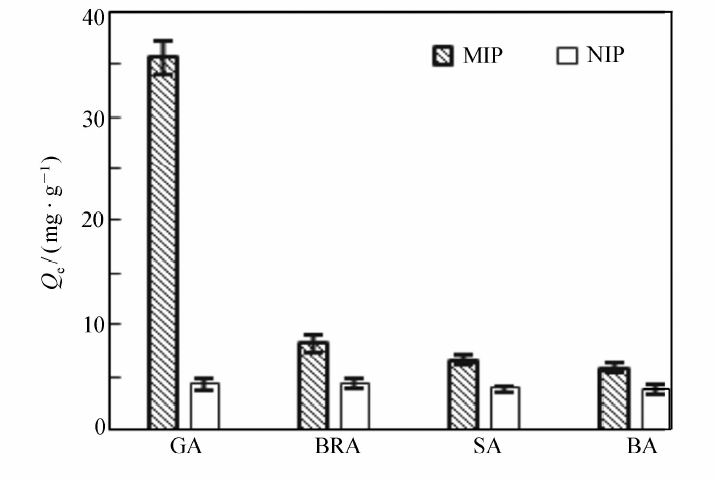 | 图8 GA为模板MIP和NIP选择性识别性能Fig.8 Selective recognition of MIP with GA as template and NIP |
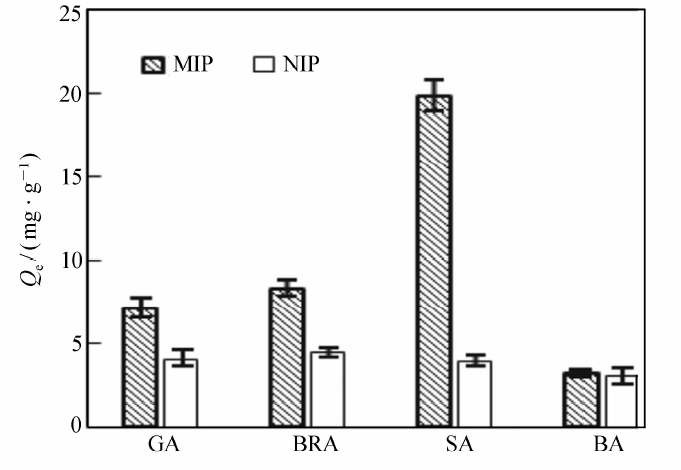 | 图9 SA为模板MIP和NIP选择性识别性能 |
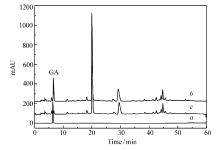
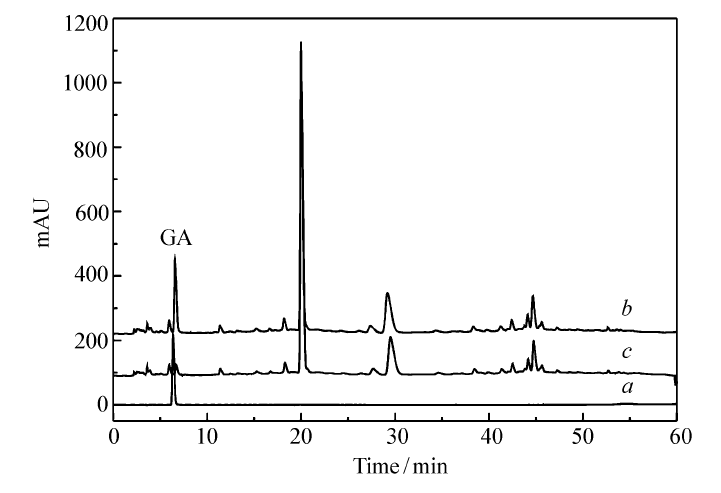
黑茶茶汤被MIP吸附前后的HPLC如图10所示。 通过GA标准溶液的HPLC图可知在保留时间约为6 min时为GA的色谱峰。比较吸附前后的色谱图发现,GA的峰面积变化很大,通过计算约有92.8%的GA已被MIP吸附,而黑茶中其它物质浓度基本保持不变,因此实验制得的MIP可从黑茶茶汤中选择性识别GA,可广泛用于GA的固相萃取。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
利用表面分子印迹技术,以纳米Fe3O4为载体,GA为模板分子、4-乙烯基吡啶为功能单体、乙二醇二甲基丙烯酸酯为交联剂、偶氮异丁腈为引发剂,成功制备了磁性表面分子印迹聚合物。 与其它印迹技术相比,由于表面印迹技术印迹只发生在硅材料载体的表面,可有效减少包埋现象与模板泄露,有利于模板分子的洗脱和识别,减少模板泄露现象,提高了模板分子的利用率。 通过TEM、FT-IR、VSM对聚合物结构和性能进行了表征,结果表明,该表面分子印迹聚合物经过双键修饰和印迹层包覆后,结构层次清楚,结合牢固,而且依然具有超顺磁性,在外磁场的作用下极易聚集;经过吸附特性研究发现,虽然MIP和NIP对GA的吸附均符合Langmuir吸附方程,为单分子层吸附,但是在相同条件下,MIP比NIP的饱和吸附量提升10倍左右,吸附性能优越;通过选择性识别性能研究发现,在相同的条件下,MIP对GA的饱和吸附量明显高于对其他羟基苯甲酸的饱和吸附量,说明该MIP对GA具有特异的选择性识别能力;将MIP应用于黑茶茶汤中GA的吸附,发现MIP可吸附92%以上的GA,完全可用于复杂体系中GA的固相萃取。 后续还需对制备条件进一步优化,不断提高核壳结构的键合牢度,提高分子印迹聚合物的重复使用次数,其应用前景将非常广阔。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|