
XU Juan, ZHAO Xinyu, KANG Congmin. Improved Synthesis of Pyrazolo[1,5-a]pyridine- 3-carboxylic Acid Derivatives[J]. Chinese Journal of Applied Chemistry, 35(5): 526-531


以取代吡啶为原料,在羟胺- O-磺酸的作用下,得到取代的 N-氨基吡啶的硫酸盐,再通过1,3-偶极环加成反应,与丙炔酸乙酯生成吡唑[1,5-a]并吡啶-3-羧酸乙酯衍生物,然后在质量分数30%的NaOH水溶液作用下水解成酸。 该方法将取代的 N-氨基吡啶的硫酸盐直接投入到下步反应,省去传统方法中将硫酸盐转化为碘盐的步骤,解决了碘盐不易析出的问题,并将取代的 N-氨基吡啶硫酸盐和丙炔酸乙酯分别用水和二甲基甲酰胺溶解后再混合,增加了原料和K2CO3在体系中的溶解性,提高了产率。 本文成功合成了6种化合物(4a~4f),产率为88%~93%,该方法条件温和,后处理简单,成本低,是适合大规模生产的新工艺。
Pyrazolo[1,5-a]pyridine-3-carboxylate derivatives were obtained from 1,3-dipolar cycloaddition reaction with ethyl propionate and N-aminopyridine sulfates synthesized by the reactions of substituted pyridines and hydroxylamine- O-sulfonic acid. They were further treated with 30%NaOH aqueous solution to give corresponding substituted pyrazolo[1,5-a]pyridine-3-carboxylic acid derivatives. In this method, N-aminopyridine sulfates were directly put into the next reaction, which eliminated the step of converting sulfate into iodine salt in the traditional method and solved the problem that the iodine salt was difficult to precipitate. N-aminopyridine sulfate and ethyl propionate were dissolved in water and N, N-dimethylformamide, respectively, and then mixed to increase the solubility of reactants. Six compounds(4a~4f) were successfully synthesized with yields of 88%~93%. The method is mild, easy to process and low cost as a new process for mass production.
吡唑并吡啶类化合物是近年来研究较多的一类稠杂环化合物,也是一类重要的反应中间体。 其中,吡唑[1,5-a]并吡啶-3-羧酸衍生物就是一类非常重要的中间体。 研究[1,2,3]表明,该类化合物在药理学方面作用显著,具有抗真菌[4]、除草[5]、杀菌[6]、抗炎症、抗焦虑、抗血栓形成、抗低血压、抗过敏、抗疟疾、止痛[7]等诸多活性,是一类研究价值很高的化合物。
吡唑[1,5-a]并吡啶骨架的合成方法主要有两大类,一类是在吡唑环的基础上合成吡啶环,该类方法比较困难,更常用的另一类是以吡啶环为基础,通过1,3-偶极环加成反应,合成吡唑环。 近几年来,报道较多的是以 N-氨基吡啶盐为前体,与碘代烯烃化合物[8]、末端烯烃化合物[9,10,11]反应。 对于吡啶上氮的胺基化反应,常用的胺化试剂有2,4,6-三甲基苯磺酰羟胺(MSH)、2,4-二硝基苯肼(DNPH)[12]和羟胺- O-磺酸[13]。 其中,羟胺- O-磺酸法收率较高,反应时间短,工艺条件简单。因此,本文选用羟胺- O-磺酸作为氮胺化试剂。
羟胺- O-磺酸法通常是先将 N-氨基吡啶的硫酸盐转化成碘盐,低温析出,然后在二甲基甲酰胺(DMF)/K2CO3体系中进行1,3-偶极环加成反应,该法中,碘盐很难析出,并且K2CO3在体系中溶解度小,目标产物收率低。 本文合成方法省去了传统方法中将硫酸盐转化为碘盐的步骤,将 N-氨基吡啶的硫酸盐纯化后直接投入到下步反应,并将 N-氨基吡啶的硫酸盐和丙炔酸乙酯分别用水和DMF溶解之后再混合,
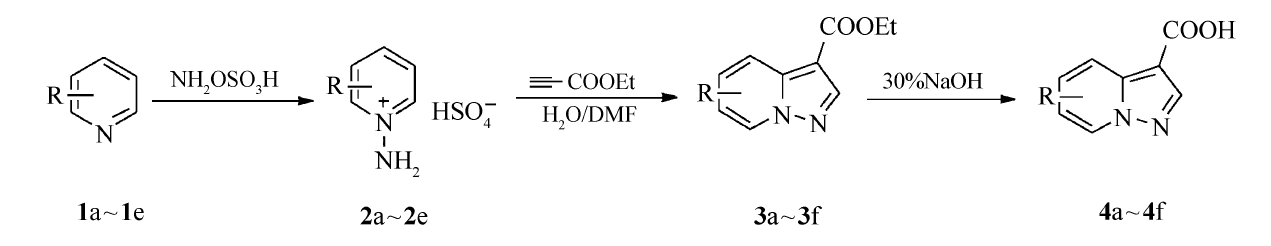 | Scheme 1 Synthetic route of the titled compounds 1a,2a:R=H; 1b,2b:R=3-CH3; 1c,2c:R=4-OCH3; 1d,2d:R=2,3-CH3; 1e,2e:R=2-OCH33a,4a:R=H; 3b,4b:R=6-CH3; 3c,4c:R=5-OCH3; 3d,4d:R=6,7-CH3; 3e,4e:R=4-CH3; 3f,4f:R=7-OCH3 |
{kind=link}
增加了原料和K2CO3在体系中的溶解性,提高了收率。 本文成功合成了6种化合物,具体合成路线如Scheme 1所示。
| 表1 标题化合物的合成 Table 1 Synthesis of the titled compounds |
其中,化合物3b和3e互为同分异构体,本文以化合物3b和3e为例,具体合成路线如Scheme 2所示。
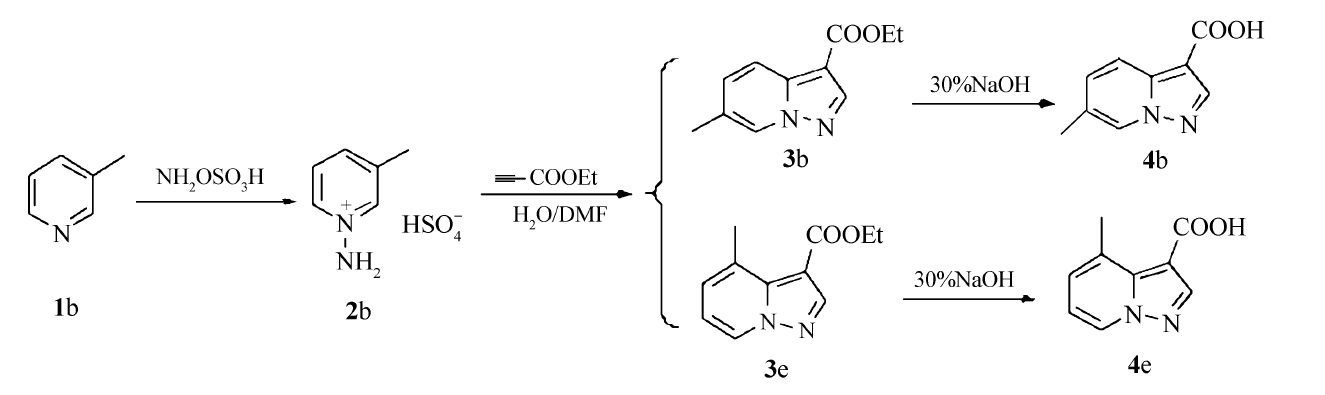 | Scheme 2 Synthetic routes of compounds 3b, 3e |
{kind=link}
XT-4型显微熔点仪(巩义市予华仪器有限责任公司);ZF-20D型暗箱式紫外分析仪(巩义市予华仪器有限责任公司);Bruker NMR500型核磁共振谱仪(瑞士Bruker公司)。
薄层层析(TLC)硅胶板(50×100 mm,青岛海洋化工有限公司);DNPH(国药集团化学试剂有限公司);吡啶,2-甲氧基吡啶,3-甲基吡啶,2,3-二甲基吡啶,4-甲氧基吡啶,羟胺- O-磺酸,丙炔酸乙酯,均购于上海达瑞精细化工有限公司;所用试剂均为分析纯。
在三口瓶中加入冰水混合物和羟胺- O-磺酸(0.01 mol),向该混合液中加入3-甲基吡啶(0.02 mol),缓慢升温至90 ℃,搅拌下反应2 h。 反应液冷却至室温,搅拌下加入K2CO3(0.01 mol),过滤,滤液旋蒸,所得固体用乙醇多次溶解,直至固体不再减少,过滤,滤液旋蒸,得到浅棕色固体。 收率90%,mp 183~185 ℃。1H NMR(500 MHz,D2O), δ:8.91(d, J=7.0 Hz,1H,ArH),8.69(d, J=7.0 Hz,1H,ArH),8.67(s,1H,ArH),8.40(t, J=7.0 Hz,1H,ArH),7.81(s,2H,NH2),2.44(s,3H,CH3)。
1.3.1 方法一
在三口瓶中加入化合物2b(0.01 mol)和水(20 mL),溶解,搅拌下,将丙炔酸乙酯(0.01 mol)缓慢滴加到三口烧瓶中,然后加入K2CO3(0.02 mol),室温下搅拌反应2 h,过滤,乙酸乙酯萃取,有机层用饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩,柱层析(洗脱剂: V(石油醚(PE)): V(乙酸乙酯(EA))=10:1)纯化,分别得到淡黄色固体产物3b和3e。 化合物3b的产率为12%,化合物3e的产率为8%。
1.3.2 方法二
将方法一中的溶剂水改为DMF,其余操作不变,得到淡黄色固体产物3b和3e。 化合物3b的产率为20%,化合的3e的产率为12%。
1.3.3 方法三
在三口瓶中加入化合物2(0.01 mol)和水(20 mL),溶解得溶液1,另取丙炔酸乙酯(0.01 mol),加DMF(10 mL)溶解,得溶液2,搅拌下,将溶液2滴加到溶液1中,向该混合溶液中加入K2CO3(0.02 mol),室温下搅拌反应2 h,过滤,乙酸乙酯萃取,有机层用饱和食盐水洗涤,无水硫酸钠干燥,过滤,滤液减压浓缩,柱层析(洗脱剂: V(PE): V(EA)=10:1)纯化,分别得到淡黄色固体产物3b和3e, 其中化合物3b是主要产物,产率48%,化合物3e产率为20%。
吡唑[1,5-a]并吡啶-3-羧酸乙酯(3a) 收率60%,mp 97~100 ℃;1H NMR(500 MHz,CDCl3), δ:8.52(d, J=7.7 Hz,1H,ArH),8.4(s,1H,ArH),8.11(d, J=7.7 Hz,1H,ArH),8.37(t, J=7.7 Hz,1H,ArH),6.92(t, J=7.7 Hz,1H,ArH),4.38(q, J=7.2 Hz,2H,CH2),1.41(t, J=7.2 Hz,3H,CH3)。
6-甲基吡唑[1,5-a]并吡啶-3-羧酸乙酯(3b) 收率48%,mp 99~101 ℃;1H NMR(500 MHz,CDCl3), δ:8.42(s,1H,ArH),7.80(s,1H,ArH),7.32(d, J=8.2 Hz,1H,ArH),6.92(d, J=8.2 Hz,1H,ArH),4.29 (q, J=7.1 Hz,2H,CH2),2.23(s,3H,CH3),1.34(t, J=7.1 Hz,3H,CH3)。
5-甲氧基吡唑[1,5-a]并吡啶-3-羧酸乙酯(3c) 收率64%,mp 109~110 ℃;1H NMR(500 MHz,CDCl3), δ:8.31(d, J=7.4 Hz,1H,ArH),8.29(s,1H,ArH),7.43(s,1H,ArH),6.62(d, J=7.4 Hz,1H,ArH),4.35 (q, J=7.0 Hz,2H,CH2),4.06(s,3H,OCH3),3.93(s,1H),1.40(t, J=7.0 Hz,3H,CH3)。
6,7-二甲基吡唑[1,5-a]并吡啶-3-羧酸乙酯(3d) 收率68%,mp 103~105 ℃;1H NMR(500 MHz,CDCl3), δ:8.40(s,1H,ArH),7.59(d, J=7.5 Hz,1H,ArH),7.25(d, J=7.5 Hz,1H,ArH),4.37 (q, J=7.0 Hz,2H,CH2),2.76(s,3H,CH3),2.39(s,3H,CH3),1.62(t, J=7.0 Hz,3H,CH3)。
4-甲基吡唑[1,5-a]并吡啶-3-羧酸乙酯(3e) 收率20%,mp 92~94 ℃;1H NMR(500 MHz,CDCl3), δ:8.40(s,1H,ArH),7.90(t, J=8.2 Hz,1H,ArH),7.64(d, J=8.2 Hz,1H,ArH),6.98(d, J=8.2 Hz,1H,ArH),4.32(q, J=7.1 Hz,2H,CH2),2.40(s,3H,CH3),1.36(t, J=7.1 Hz,3H,CH3)。
7-甲氧基吡唑[1,5-a]并吡啶-3-羧酸乙酯(3f) 收率60%,mp 116~119 ℃;1H NMR(500 MHz,CDCl3), δ:7.82(s,1H,ArH),7.10(t, J=8.4 Hz,1H,ArH),6.42(d, J=8.4 Hz,1H,ArH),5.92(d, J=8.4 Hz,1H,ArH),4.30 (q, J=7.2,2H,CH2),4.06(s,3H,OCH3),1.32(t, J=7.2,3H,CH3)。
在三口瓶中加入化合物3b(0.01 mol)和乙醇(10 mL),将化合物3b溶解,向该溶液中加入30%NaOH水溶液,缓慢升温至90 ℃,搅拌下反应5~6 h(TLC检测),反应液冷却到室温,用盐酸调pH=2,过滤,滤饼水洗,干燥,得白色固体产物。
吡唑[1,5-a]并吡啶-3-羧酸(4a) 收率93%,mp 217~219 ℃;1H NMR(500 MHz,DMSO- d6), δ:12.46(s,1H,COOH),8.85(d, J=7.8 Hz,1H,ArH),8.39(s,1H,ArH),8.08(d, J=7.8 Hz,1H,ArH),7.56(t, J=7.8 Hz,1H,ArH),7.13(t, J=7.8 Hz,1H,ArH)。
6-甲基吡唑[1,5-a]并吡啶-3-羧酸(4b) 收率90%,mp 223~225 ℃;1H NMR(500 MHz,DMSO- d6), δ:12.70(s,1H,COOH),8.42(s,1H,ArH),7.80(s,1H,ArH),7.32(d, J=7.2 Hz,1H,ArH),6.91(d, J=7.2 Hz,1H),2.42(s,3H,CH3)。
5-甲氧基吡唑[1,5-a]并吡啶-3-羧酸(4c) 收率89%,mp 232~235 ℃;1H NMR(500 MHz,DMSO- d6), δ:12.29(s,1H,COOH),8.70(d, J=7.5 Hz,1H,ArH),8.27(s,1H,ArH),7.32(s,1H,ArH),6.79(d, J=7.5 Hz,1H,ArH),3.90(s,3H,OCH3)。
6,7-二甲基吡唑[1,5-a]并吡啶-3-羧酸(4d) 收率92%,mp 229~232 ℃;1H NMR(500 MHz,DMSO- d6), δ:12.40(s,1H,COOH),8.41(s,1H,ArH),8.22(d, J=8.4 Hz,1H,ArH),8.10(d, J=8.4 Hz,1H,ArH),2.45(s,3H,CH3),2.02(s,3H,CH3)。
4-甲基吡唑[1,5-a]并吡啶-3-羧酸(4e) 收率88%,mp 203~205 ℃;1H NMR(500 MHz,DMSO- d6), δ:12.76(s,1H,COOH),8.50(s,1H,ArH),7.80(t, J=8.2 Hz,1H,ArH),7.41(d, J=8.2 Hz,1H,ArH),6.96(d, J=8.2 Hz,1H),2.49(s,3H,CH3)。
7-甲氧基吡唑[1,5-a]并吡啶-3-羧酸(4f) 收率88%,mp 249~251 ℃;1H NMR(500 MHz,DMSO- d6), δ:12.70(s,1H,COOH),7.82(s,1H,ArH),7.46(t, J=8.0 Hz,1H,ArH),6.98(d, J=8.0 Hz,1H,ArH),6.24(d, J=8.0 Hz,1H,ArH),4.22(s,3H,OCH3)。
绝大多数相关文献[8,9,11,12,13]中,只是给出化合物3b的合成方法,并未提及同分异构体3e,本文将化合物3b和3e进行了分离纯化,并对产物结构进行讨论。 由化合物2合成化合物3的反应属于1,3-偶极环加成反应,化合物2作为1,3-偶极体与亲偶极体丙炔酸乙酯合环,该合环过程需要吡啶上2位或6位的碳原子参与,所以当吡啶环的2位或6位无取代基时,就会有同分异构体产生。 以3-甲基吡啶为原料,2位和6位分别在甲基的邻对位,电子效应对成环的位置影响不大,成环位置主要受空间位阻的影响,所以空间位阻小的6位比2位容易成环,对应的产物3b为主产物,产率为48%,同分异构体3e为次要产物,产率为20%。
化合物2是硫酸盐,在DMF中的溶解性很差,能溶解在水中,但丙炔酸乙酯不溶于水,所以考虑用水和DMF分别溶解后再混合,为考察不同溶剂条件对产物3收率的影响(以化合物3b和3e为例说明),分别探索了只用水溶、只用DMF溶和将1-氨基-3-甲基吡啶的硫酸盐和丙炔酸乙酯分别用水和DMF溶解之后再混合3种方法,结果如表2所示。 由表2可见,水和DMF单独做溶剂时,由于反应液是两相体系,产率很低,而用水和DMF分别溶解后再混合,反应液为均相体系,产物的产率大大提高。
| 表2 溶剂对化合物3b和3e总收率的影响 Table 2 Effect of different solvents on yields of compounds 3b and 3e |
综上所述,本文以取代吡啶为原料,以羟胺- O-磺酸为氮胺化试剂,合成了 N-氨基吡啶硫酸盐,经乙醇纯化后,在H2O/DMF 的混合溶剂中与丙炔酸乙酯发生1,3-偶极环加成反应,得到取代的吡唑[1,5-a]并吡啶-3-羧酸乙酯,最后在30%NaOH水溶液作用下水解成性质更稳定吡唑[1,5-a]并吡啶-3-羧酸衍生物。 并且讨论分析了产物结构,探究了溶剂和反应时间等条件对收率的影响,优化了工艺条件。
在由取代吡啶合成吡唑[1,5-a]并吡啶-3-羧酸乙酯衍生物的路线中,对传统的合成方法进行了改进,省去了传统方法中将硫酸盐转化为碘盐的步骤,将 N-氨基吡啶的硫酸氢盐纯化后直接投入到下步反应,并将传统方法中只用DMF做溶剂,改为将 N-氨基吡啶的硫酸氢盐和丙炔酸乙酯分别用水和DMF溶解之后再混合,增加了原料和K2CO3在体系中的溶解性,提高了收率。新的合成途径条件温和,容易控制,后处理简单,产率高,成本低,是适合大规模生产的新工艺。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|