
CHEN Hang, LIU Chundong, WANG Jianhua. Research Progress on Modification and Effects of Halogen on Nucleic Acid Drugs[J]. Chinese Journal of Applied Chemistry, 35(5): 491-499


药物分子中引入卤素具有改善药物脂溶性、影响药物分子的电荷分布及影响药物的作用时间等特点,故被广泛应用。 作为抗病毒、抗肿瘤、干扰素诱导和免疫增强的、具有特定碱基序列的核酸药物也不例外。 本文综述了卤素对核酸结构中的嘧啶类、嘌呤类和核糖结构修饰后的核酸类药物生物活性产生的影响,以期为核酸及核酸类似物的药物研发提供新的思路。
The halogenation is an invaluable approach for structural modification of drugs in medicine chemistry development. It increases the lipo-solubility, influences on the space charge distribution and improves their drug metabolic activity. Nucleic acid, as antiviral agent, antitumor agent, interferon inducer and immunopotentiator, can also be modified by halogenation. In this review we will discuss the effect on the bioactivity of modified nucleic acids on pyrimidines, purines and ribose by halogenation, and summarize the latest developments in drug research and development of modified nucleic acid.








核酸是生命中最基本物质之一,一般由碱基(嘧啶类、嘌呤类)、核糖和磷酸组合而成。 从20世纪四五十年代起,人们开始研究核酸的生物活性,取得了很大的成果,如明确了核酸的结构及其在细胞中的作用。 随着对核酸代谢过程的了解,人们开始研究核酸衍生物。 在药物化学中,将生物分子用卤素(Cl、Br、I和F)修饰是一个研究的热点。 大约有40%的卤化药物进入市场或临床前试验阶段,迄今有大约25%的有机卤化药物进入市场,而34%的卤化药物仍处于研发阶段[1],这表明卤素在药物研发中起着重要的作用。 其中,氟和氯在药物化学中广泛应用,而含碘的药物应用很少[2]。 但目前相对于有机氟,相对分子质量更大的有机卤素在药物研发中呈上升趋势,为研发新的先导化合物提供了新的思路[1,2]。 在美国国家癌症中心(National Cancer Institute's Diversity Set IV)所批准的高通量筛选小分子抗癌先导化合物中,有20%的分子包含I、Br和Cl[1]。 卤素具有增加分子亲脂性、提高对脂质膜渗透性以及卤素的电负性可以增加中心分子生物活性等特点[3]。 例如,引入强吸电子基团如F可以增强结合作用、代谢稳定性、物理特性的变化以及选择活性[4]。 此外,还有文献报道,通过卤素对嘧啶环、嘌呤环和核糖的修饰,使得核酸衍生物表现出更强的抗病毒、抗代谢、抗菌等特性[5]。 由于卤化的核酸衍生物所具有的较好生物活性,目前已报道了大量卤素修饰核酸的方法。 本文拟对卤素在核酸中嘧啶类、嘌呤类和核糖等结构中的修饰与活性进行简述。
嘧啶核苷是一类芳杂环,包括尿苷,胞苷和胸苷。 嘧啶核苷不但可用于合成核酸(包括从头合成和补救途径两种方式合成),而且还是糖和脂质代谢所必需的[6]。 如尿苷二磷酸(UDP)-葡萄糖、胞苷二磷酸(CDP)-二酰基甘油和CDP-胆碱分别为合成糖原、甘油磷脂的活性中间物。 由于嘧啶类在生命进程中起着重要的作用,故通过卤素对嘧啶类的修饰以改善其生物学活性。
由于嘧啶可以合成核酸,通过卤素对嘧啶的修饰使其影响逆转录酶、DNA聚合酶等活性,破坏细菌,病毒正常合成的过程,从而具有抗菌、抗病毒等活性。 Ono等[7]对1- β-D-阿拉伯呋喃糖尿嘧啶-5'-三磷酸盐(aUTP)进行C-5位F、Cl、Br和I修饰,以aUTP为对照,研究4种卤代衍生物对小鼠细胞及RNA病毒中DNA聚合酶活性的影响。 结果表明,尿嘧啶卤代衍生物的诱导效应、空间位阻、疏水作用对酶抑制作用会有影响。
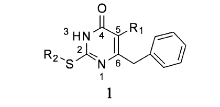
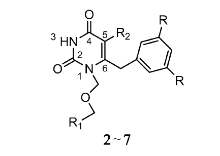
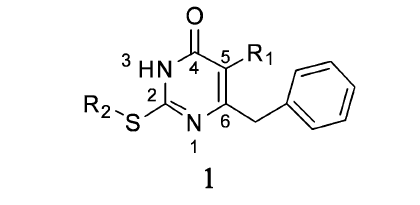
在卤素修饰中,相对分子质量较大的卤素在研究过程中呈上升趋势,这归因于其良好的生物学活性。 Qin等[8]对嘧啶衍生物 S-二氢烷氧苄基嘧啶酮(S-DABOs)(1)(如图1)进行R1、R2修饰,通过比较在R1上不同的取代基,实验表明甲基取代比H活性稍微高一些,而用Br或I取代,其活性明显比H和甲基高。 由半数抑制浓度(IC50)可知,I活性比Br高6倍甚至更多。 例如,若R2为萘甲基,卤素在R1的取代是影响其活性的主要因素。 结果I取代的化合物(IC50=2.04 μmol/L)活性明显比Br取代高(IC50=12.26 μmol/L),说明抑制HIV-1RT病毒活性与卤素原子的亲电性及原子半径有关。 又如,当R2为C6H5CH2CH2CH2—时,R1由I取代(IC50=0.18 μmol/L)活性比由H取代(IC50=8.17 μmol/L)高45倍,比奈韦拉平(IC50=4.12 μmol/L)高23倍。 通过结构活性关系分析S-DABOs在R1的取代,活性顺序为I>Br≫Me-H。 可见,这对治疗HIV-1具有很大的应用潜力。 ŽupanČić等[9]使用5-卤嘧啶衍生物对6种人类癌细胞进行评价。 这些化合物中,只有5-碘尿嘧啶衍生物对人类结肠癌细胞有中等强度抑制作用。 由于耐药突变体的出现限制了非核苷逆转录酶抑制剂(NNRTIs)的功效,因此必须开发具有更好的耐药性和药代动力学特征的新型抗病毒药物。 Wang等[10]在抗HIV-1病毒药物的研究中,通过对1-[(2-苄氧基/烷氧基)甲基]-5-卤-6-芳尿嘧啶的修饰,得到的化合物2、3、4、5和6(见图2,表1)在野生型HIV-1和耐NNRTIs的HIV-1链中表现出显著的抑制活性。 它们对HIV-1 RT的IC50分别为0.110、0.003、0.043、0.021和0.015 μmol/L。 化合物3,具有最高的选择性指数(SI=38215),比奈韦拉平和TNK-651更有潜力。 研究表明,在C-5位引入卤素可能会增加化合物对逆转录酶异变体的抑制作用。 例如,嘧啶环上R2被卤原子特别是I取代,会增加化合物与负电子基团的相互作用。 如化合物7 (见图2,表1)与络氨酸188的羰基氧的相互作用,I取代使各向异性的表面电荷具有显著正电荷的特性,表现出路易斯酸的性能。 因此,在嘧啶环上引入吸电子基团可能增加与络氨酸188羰基氧电子间的相互作用。 此外,I与羰基氧的距离(0.335 nm)低于范德华力作用的距离(0.355 nm),化合物7形成的C—X••••O键角157.93°与预计的理想键角165°很接近。 在C-6位上引入二甲基芳香基团可以显著增强抑制耐NNRTIs药物的HIV-1链的活性。 如果用二氟取代物取代二甲基,对HIV-1仍具有很高的抑制活性。 这可能是因为氟的引入有利于改善缺电子的苯环与富电子的络氨酸188苯环所形成的 π堆积相互作用。 Yan等[11]也是通过对芳基嘧啶进行卤素修饰而得到新的NNRTIs药物,在抑制野生HIV-1方面取得显著效果。
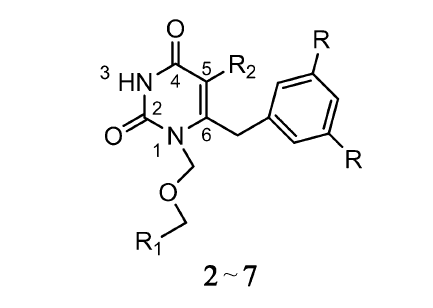
| 表1 1-[(2-苄氧基/烷氧基)甲基]-5-卤-6-芳尿嘧啶不同取代基修饰 Table 1 Different preparation of 1-[(2-benzyloxy/alkoxy) methyl]-5-halo-6-auracil |
二芳基嘧啶(DAPY)衍生物中的依曲韦林和利匹韦林作为抗HIV-1的NNRTIs具有很好的潜力。 Yan等[11]早期对DAPY进行了结构修饰,得到的CH(OH)-DAPYs衍生物中,发现引入小分子后效果增强,而卤素的引入有利于配体与蛋白的结合,故合成了一系列新的含卤DAPY衍生物,并评估其在MT-4细胞培养物中的抗HIV活性。 其中化合物8、9(如图3)对于野生型HIV-1的半最大效应浓度(EC50)分别为0.005和0.009 μmol/L。 特别是化合物9,对双突变体103N+181C有很强的抑制活性,其EC50为8.20 μmol/L。

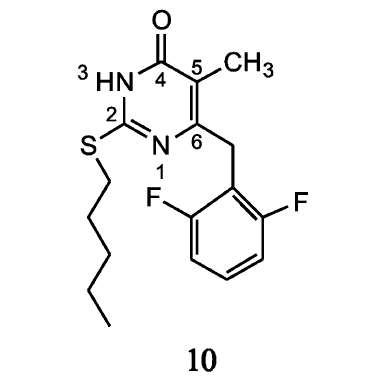
通过对二氢烷氧苄基嘧啶酮(DABOs)如S-DABOs进行分子模拟设计,得到的2,6-双取代苯甲基-DABO衍生物能够高效特异地抑制HIV-1逆转录酶。 为了进一步得到其它的DABO衍生物,Antonello等[12]研究了吸电子基团对取代S-DABO骨架的苄基后抗HIV-1活性的影响。 当对S-DABOs中嘧啶环的C-6苯环进行2,6位双卤素取代时,结果对HIV-1抑制作用相比于其它吸电子基团效果更强。 不同的单取代苯及双取代苯化合物被合成,在细胞和重组逆转录酶中作抗HIV-1试剂来评价其效力,这些化合物中最有潜力的是6-(2,6-二氟苯基)取代(F-DABOs)(10)(如图4),其EC50为0.04~0.09 μmol/L,选择性指数(SI)≥5000。 正如Wang等[10]提到,氟的引入有利于改善缺电子的苯环与富电子的络氨酸188苯环所形成的 π堆积相互作用。 从而提高对HIV-1逆转录酶的抑制作用。 Metwally等[13]在苯环引入卤素显著增加了对癌细胞的作用。 可见,卤素与苯环之间可能因为构效关系而产生一定的活性,这为我们抗病毒药物的设计提供了思路。
尽管如此,卤素和苯环的引入也可能会产生细胞毒性。 如Kumarasamy等[14]合成了4-取代3,4-二氢嘧啶-2衍生物,(如图5),化合物11具有长的亲脂性侧链,有利于选择性抑制Punta Toro病毒。 但若R基团由含有卤素的芳基取代,对抗病毒没有明显的作用,相反,会增加对MT-4细胞的毒性。

腺苷是一种天然存在的核苷,涉及多种生理和病理生理过程。 其一些生理作用包括对心率和心房收缩力的影响,血管平滑肌张力,神经递质的释放,血小板功能,脂肪分解,肾功能和白细胞功能[15]。 通过对嘌呤的修饰以改善其生物学功能,其中,卤素的修饰是一个重要的研究方向。
Endo等[16]合成的8-溴-2-辛炔-N9-炔腺嘌呤作为腺嘌呤A2A受体拮抗剂,对帕金森疾病的小鼠模型IC50为(0.0051±0.00032) μmol/L。C-8位由氯,2-呋喃环取代也表现出很好的抑制活性,其IC50分别为(0.0060±0.00050) μmol/L和(0.0036±0.00065) μmol/L。 Yin等[17]也对C-8位进行修饰,形成的C-8碘-氮杂-脱氧鸟苷,与对照组相比,经碘-氮杂-脱氧鸟苷处理后的HeLa细胞减少了80%,表现出良好的抗恶性细胞增殖的能力,具有抗癌药物的潜力。
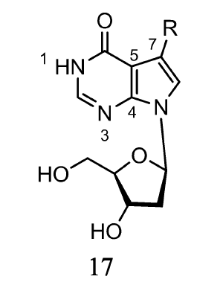
吡咯并[3,2-d]嘧啶即9-氮杂嘌呤,由于其与嘌呤具有相似性,因而得到广泛的研究。 Temburnikar等[18]早期研究发现,经卤素修饰的吡咯并嘧啶化合物对三阴性乳腺癌细胞系(TNBC)MDA-MB-231的影响与不含卤素的化合物有差异。 为了进一步探究卤素对该化合物的影响,该实验室合成了一系列卤素取代的吡咯并嘧啶化合物,其中,化合物15(如图6)对恶性细胞如L1210 CEM HeLa的IC50分别是(6.8±2.8) μmol/L、(25±2) μmol/L和(19±3) μmol/L,而在C-7位引入碘(16)可以很大程度增加抑制活性,对3种细胞的IC50分别为0.93 μmol/L、(4.9±0.4) μmol/L和(0.92±0.04) μmol/L。 而将这两种化合物用于研究三重阴性乳腺癌细胞MDA-MB-231,化合物15在G2/M阶段诱导细胞凋亡效果不明显,而化合物16很大程度抑制细胞凋亡。 有研究表明,化学不稳定部分作为亲电共价抑制剂会产生脱靶效应,效果不令人满意。而卤素形成的共价键具有不可逆的抑制作用,使其在癌症治疗方面提供新的方法。

卤素在碱基配对方面也有影响。 Seela等[19]对化合物17(如图7)C-7位进行卤素和炔基取代,结果表明,这些修饰后的化合物与7-脱氮-2'-脱氧肌苷(c7Id)和2'脱氧核苷类似,在与A、T、C、G配对中表现出非特异性配对(模糊配对),而与碱基形成的双链稳定性随着Cd>Ad>Td>Gd递减。 将5'-7-氮杂-7-卤-2脱氧核苷酸-C)6即5'-d(F7c7I-C)6-3'( Tm=28 ℃)、5'-d(Br7c7I-C)6-3'( Tm=33 ℃)和 5'-d(I7c7I-C)6-3'( Tm=27 ℃)进行自身配对,其稳定性比母链5'-d(c7I-C)6-3'( Tm=13 ℃)高。 我们实验室对核酸类似物肽核酸也进行了类似的研究。 结果表明,经卤素修饰的胸腺嘧啶(5-XU):A稳定性比T:A高[20]。 非特异性配对表现出良好的稳定性有利于在生物研究过程中获得需要的突变,这使得它们在核酸化学、分子生物学及纳米生物技术中作为探针有广泛的应用[19,21]。

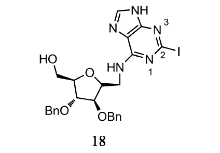
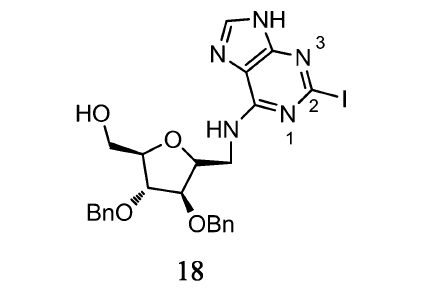
目前,卤素与药物分子形成卤键也是一个重要的研究方向。 Ramzaeva等[22]通过X射线分析了C-7位由卤素(Br,I)取代的Br7c7z8Gd、I7c7z8Gd等核酸衍生物晶体结构,发现C-7卤与相邻核苷的N-3之间形成卤键。 卤素本身带负电,但是其头部由于电子的缺少,会有一个正静电势区域— σ-hole。 由于 σ-hole存在于卤素原子的头部,而卤素修饰的N-3原子显示部分负电荷性质。 这种新的键使卤素修饰的核酸衍生物表现出很高的稳定性。 此外,有研究报道,通过卤素对嘌呤衍生物C-2位修饰,有利于其表现良好的生物学活性。 如Tak-Tak等[23]制备的C-2位由碘取代的嘌呤衍生物(18)(如图8)对FGFR3激酶的抑制作用最强,通过分子对接实验表明,由碘形成的卤键在相互作用中起关键作用。 目前,卤键已广泛用于各种化学领域,如超分子、生物和无机化学[24,25,26]。 卤键的强度与很多因素有关,如卤素的极性、与卤素共价结合的原子的电子云密度等[27]。 对于卤素而言[28],其卤键强度遵循Cl<Br<I。 这为设计核酸形成卤键提供了理论基础。
Zhao等[4]早期在开展嘌呤类物质抑制葡萄球菌研究中,发现噻唑并[3,2-a]嘧啶-3-酮衍生物中R2(如图9)是药效基团。 但为了改善该化合物的抑菌性能,该实验室对化合物进行了进一步修饰。 在临床中,由于氯在抗分枝杆菌方面应用广泛,因此,他们猜测氯和电子云密度更大的氟有可能会增加化合物结合能力,改善物理性能及选择活性。 通过检测合成的一系列化合物,结果表明,由卤素取代该衍生物得到的化合物19和20(如图9),对葡萄球菌和金黄色葡萄球菌比标准药物头孢唑林表现出更强的抑制活性,MIC值在1.56~6.25 μmol/L范围内。 此外,该化合物对于抑制YycG组氨酸激酶有着明显的活性。
 | 图9 卤代噻唑并[3,2-a]嘧啶-3-酮羧酸[4]Fig.9 Halogenatedthiazolo[3,2-a]pyrimidin-3-onecarboxylic acid derivatives[4] |
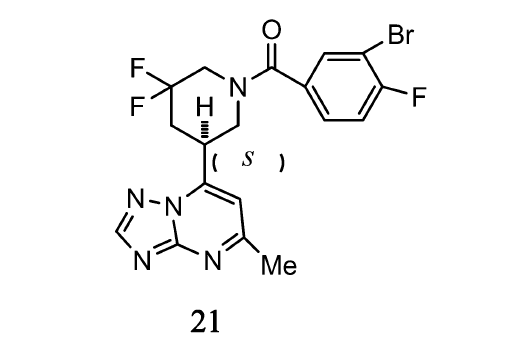
第二信使cGMP和cAMP在突簇可塑性和记忆过程中具有重要作用。 而通过对PDE2a酶的抑制,可以调节环核苷酸在细胞内的浓度,对于记忆力下降或者其他的认知缺陷具有潜在的治疗意义。 Gomez等[29]通过对药物结构的设计以及分子模拟,得到了一系列有效且可选择性抑制PDE2a酶的化合物。 其中, S构型的先导化合物21(如图10)对于PDE2a酶有很高的选择性且抑制效果明显,该化合物在小鼠体内有很好的药动学特征。 其IC50为0.008 μmol/L,小鼠体内血浆清除率为25.7 μL/(min·mg),药物在血脑屏障的渗透率为20.7×10-6 cm/s。 这主要是由于化合物的卤素与Tyr827的氧在PDE2a的活性位点形成卤键,导致对PDE2a酶的抑制作用增强。 因此,该化合物可作为研究选择性抑制PDE2a酶的工具。
随着人们对HIV、乙肝病毒(HBV)等病毒的研究,目前对核苷酸药物的修饰有了深入的认识。 其中,在糖基上通过对2',3'不饱和双脱氧修饰有利于抗病毒作用,如司他夫定。 而用氟糖分子取代核苷环越来越引起研究者注意,它们中很多化合物都具有抗病毒抗癌的活性[30]。 此外,在2'位引入氟有利于加强糖基键的稳定性,表现出不同的生物活性。 这个性质在2'-脱氧核糖嘌呤核苷酸和4'-脱氧核糖嘌呤核苷酸中最明显,因为它们在酸性介质中不稳定,口服影响其生物活性。 而2'- β-氟-2',3'双脱氧腺苷酸及2'-氟-2',3'不饱和嘌呤均表现出抗HIV的活性。 有研究表明,对氟原子的引入有利于改变化合物的物理、化学、生物学特征[30]。
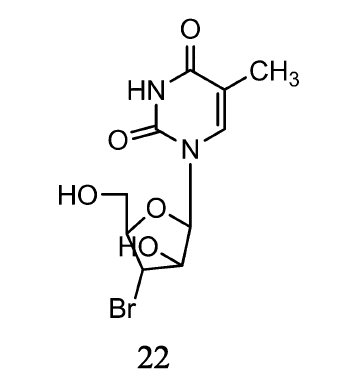
Shakya等[31]合成了由2'-或3'-卤代的嘧啶核酸衍生物,如尿嘧啶、5-氟尿嘧啶以及胸腺嘧啶,并且评价了其抗结核分枝杆菌的活性。 其中,3'-溴-3'-脱氧-阿糖胸苷(22)(如图11)在体外评价中对结核分枝杆菌抑制作用最强。 该化合物对野生型结核病菌株(H37Ra)的MIC50为3.14 μmol/L,对耐利福平和异烟肼菌株(H37Rv)的MIC50为3.14~6.28 μmol/L。 该化合物对细胞内感染有H37Ra的人单核细胞系(在31.1 μmol/L浓度下减少80%)显示出比细胞外更高的抗分枝杆菌活性(在31.1 μmol/L浓度下减少75%)。 3'-卤素取代基活性顺序为Br>Cl>F=I。 但2'-溴取代物在抗结核分枝杆菌研究中活性减弱甚至无活性。
丙型肝炎(HCV)属于分布很广的疾病,一些抑制HCV的核苷酸类似物已有报道。 通过进一步对这些核苷酸类似物糖基修饰,包括3'-脱氧核苷酸,2'-C-CH3,2'- α-F,2'-脱氧-2'- α-F -β-C- CH3,2'-O-CH3核苷酸,4'-氮杂胞苷等以及对杂环碱基的修饰,得到了一系列新的抑制HCV化合物。 此外,由氟修饰的核酸在抑制病毒(包括HCV)复制方面应用十分广泛,故Ivanov等[32]新合成了含氟的核酸衍生物—4'-氟尿嘧啶-5'- O-三磷酸盐(23)(如图12),其表现出对HCV依赖的RNA聚合酶有抑制作用(IC50=2 μmol/L)。

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
由于卤素具有很强的电负性,可以改善化合物活性,有利于提供电子形成π堆积相互作用;亲脂性有利于化合物进入细胞;诱导作用加强化合物生物活性等一系列优势,使得卤素在药物合成中具有广泛的应用。 此外,在药物分子设计中,在卤素与分子间相互作用形成卤键方面得到了广泛的研究[33,34,35]。 探究二者形成卤键后的一系列性质,这也是目前研究的主要方向。 由于卤原子取代氢原子的质量效应以及卤素的电负性,5-卤尿嘧啶中分子环呼吸振动及凯库勒伸缩振动比尿嘧啶振幅更低,且C-4=O的共振相比于尿嘧啶发生偏移[36]。 此外,卤素的引入还改变了嘧啶其它性质,如偶极矩的大小和方向。 这些性质可能会诱导化合物构象的改变,但C-5位卤代产生的影响比C-6位小[37]。 一线抗癌药物5-氟尿嘧啶,作为抗肿瘤代谢药物广泛用于临床。 由于C-5位是活性位点,目前研究报道中,卤素对嘧啶及嘧啶衍生物修饰都主要是基于此合成的。 由于氟原子半径和氢原子相近,且形成的C-F键特别稳定,故5-氟尿嘧啶能以分子形式参与正常代谢。 但通过用不同取代基对嘧啶环进行结构修饰,使得C-5位并非只有F才具有活性。 如C-6位由苯环取代时,苯环经卤修饰后,其生物活性较好。 相反,C-5位的卤素取代对化合物的影响甚至不是很明显。 对于嘌呤,卤素修饰的活性位点主要是C-2、C-7、C-8,而卤素对核糖的修饰主要为2'、3'。 通过形成卤键、改变化合物脂溶性、p Ka等因素使其表现出显著的生物活性。 此外,经卤素修饰的碱基有利于碱基的互补配对,提高核苷酸双链稳定性。 但由于卤素的引入可能会引起化合物的构型、空间位阻等改变,因而在引入卤素时需要考虑卤素原子的大小,引入后化合物活性的变化。 此外,活性位点的修饰是很重要的,否则可能会产生细胞毒性。
有研究表明[38,39],体内的次氯酸(HOCl)能与嘧啶反应,如生成5-氯尿嘧(ClU)、5-氯胞嘧啶(ClC)。 如果最初是以A-T配对,那么可能会产生ClU-A这样的错误配对。 用NMR检测,ClU-A与A-T几何结构具有相似性。 这个模拟的沃森-克里克ClU-A碱基对是足够稳定的,不受糖基酶移除,因此在细胞内可能构成持续损伤DNA的形式。 而次氯酸介导的损伤常在炎性位点发现,表明它与炎症及人类的疾病,甚至包括癌症有关。 因而,如果用卤素修饰碱基,是否会对机体产生类似的反应还需要进一步研究。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|