

GUAN Xiaolin, MENG Li, JIA Tianming, et al. Synthesis and Electroluminescent Properties of a Liquid Crystalline Linear Conjugated Arylacetylene Derivative[J]. Chinese Journal of Applied Chemistry, 35(4): 426-435


以苯胺和芳炔为基本构筑单元,通过Sonogashira偶联反应, 合成了一种新型含氨基的结构不对称 π共轭线性芳炔化合物5-{(6-己氧基萘基)丁二炔}-2-{(4-氨基苯基)乙炔}苄醇。 通过化学修饰在芳炔类小分子端基引入氨基取代基,使其在无吸电子基团存在的条件下通过扭转态的形成实现分子内电荷的有效转移,从而提高芳炔类衍生物电-光转换效率。 同时,通过赋予芳炔类小分子液晶性,有效改善电子与空穴在器件中的电荷平衡,提高器件的效率。 基于氨基取代芳炔衍生物为掺杂发光材料制备的电致发光器件呈现黄绿光发射,器件开启电压较低(7.20 V),显示了较好的电致发光稳定性,器件17.65 V时达到最大亮度126 cd/m2,是一种潜在的电致发光材料。
A novel asymmetrical linear conjugated diacetylene derivative, (2-(2-(4-aminophenyl)ethynyl)-5-(4-(2-(hexyloxy)naphthalen-6-yl)buta-1,3-diynyl)phenyl)methanol, was designed and synthesized from phenylamine and arylacetylene by Sonogashira reaction. By attaching the phenylamino substituent to arylacetylene, torsional states will be formed under conditions of the absence of electron withdrawing group and an effective intramolecular charge-transfer occurs, which helps improve electro-optical conversion efficiency. Furthermore, the introduction of liquid crystal property improves the charge balance of electron and hole in device and device efficiency. Moreover, an organic light-emitting device(OLED) device using the compound as emitters was fabricated, which exhibited yellow-green emission and low turn-on voltages of 7.2 V and excellent electroluminescent stability. The maximum brightness value was 126 cd/m2. So, the diacetylene derivative is a promising materials for OLED devices.




有机发光二极管(OLED)显示器件的产业化是21世纪信息产业中一个重要组成部分,与其它显示器件相比,其具有响应速度快、驱动电压低、能耗小、结构简单、易于实现高分辨和宽视角显示、环境适应性强、易实现全色大面积显示等优点,使其广泛应用于电视、计算机、数码相机、车载显示,军事与航天等许多领域,是当前国内外的研究热点[1,2,3,4]。 高性能OLED器件制备的关键在于光电功能材料的开发和选择,而发光材料光学性质是决定器件性能的重要因素之一,这使得设计合成具有实际应用价值的新型有机电致发光材料成为OLED发展的基础和关键。 而有机小分子光电材料因具有结构可调,易于合成、稳定性好及成本较低等优势,引起了广泛的关注和研究[5,6,7]。
有机光电材料通常是含有 π共轭体系和氮、硫等杂原子的芳香性有机分子,分为小分子化合物和聚合物两类。 在众多的有机小分子光电材料体系中,芳炔类化合物因含有大的共轭π电子体系、强的刚性结构和高荧光量子产率,被广泛用作荧光材料、液晶材料(LC)、有机半导体材料和OLED等[8,9,10,11,12,13,14,15]。 芳炔类化合物结构中的各类芳烃往往通过炔基( -C≡C-)桥联在一起,具有将电子从 π-共轭体系的一端传送到另一端的能力,因此该类化合物拥有较好的电子输送效应。 同时,芳炔类化合物的摩尔吸光系数(ε)和最大吸收波长( λmax)可通过改变芳香基团和共轭长度来调节,因此可作为高效光电材料[16]。 近年来,含各种官能团的芳炔类衍生物已被合成。例如,Flatt等[17]合成了端基含有乙酰巯基的苯乙炔类荧光分子线。 Fasina等[18]合成了末端含羧基的共轭芳炔化合物并对其光谱性质进行了研究。 李海涛课题组[19]合成了一系列新型的具有良好光、电化学响应性的氨基封端的共轭芳炔化合物。 相比于其它官能团,氨基基团取代的芳炔类荧光分子在激发态下会形成扭转型分子内电荷转移(TICT)态[20]。 Hirata等[21]通过研究含氨基的芳炔化合物的发光情况,证明此类化合物可以在无吸电子基团存在的条件下通过扭转态的形成实现分子内电荷的有效转移。 因此,氨基取代的芳炔小分子有望成为一种高效OLED发光材料。
此外,普通小分子荧光材料仍然面临电致发光效率较低,且电-光转换效率差的问题。 而有研究报道,含大共轭结构的液晶材料因同时具有液体的流动性和晶体的有序性,将液晶基元引入到载流子传输材料中,能有效改善电子与空穴在器件中的电荷平衡,大大提高器件的效率[22,23]。 我们前期工作也证实了具有液晶性的线性共轭不对称芳炔化合物具有良好的电致发光性能,启动电压较低,发光稳定性较好,可作为一种电致发光材料使用[24]。 因此,赋予氨基取代芳炔类小分子液晶性将有助于提高此类材料的电-光转换效率。 而据我们所知,具有液晶性的基于氨基芳炔衍生物在电致发光器件领域的应用研究尚不多见。
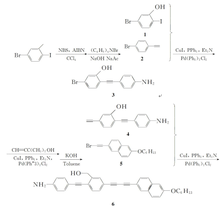
本文综合考虑液晶性对提高器件效率的作用,以及氨基芳炔类小分子荧光材料优良的光电性质,并结合此前的研究,设计并合成了分子结构中包含芳炔和苯胺的线性共轭不对称液晶小分子,研究了分子的液晶相行为,探讨溶剂化效应对其发光性能的影响。 同时,对此种新型高共轭液晶材料,采用旋涂的方式制备小分子器件,研究了液晶分子的电致发光性能。 这也是对具有液晶性和TICT态有机小分子电致发光器件研究的一次新尝试。 合成路线如Scheme 1所示。
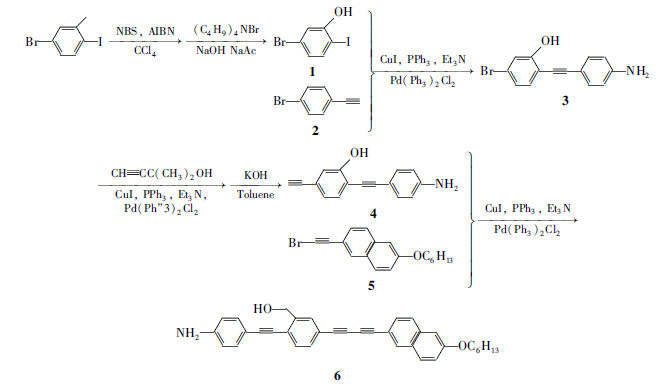 | Scheme 1 Synthesis of liquid crystal compound 6 |
{kind=link}
对溴苯胺(4-Bromoanilines,分析纯)、碘化亚铜(CuI,≥98%),Zn粉(分析纯)、三苯基磷(PPh3,≥98%)、双三苯基磷二氯化钯([(C6H5)3P]2PdCl2,≥99.95%)、2-甲基-3-丁炔-2-醇(2-Methyl-3-butyn-2-ol,≥97%)、5-溴-2-碘甲苯(C7H6BrI,≥98%)、1-溴代正己烷(1-Bromohexane,分析纯)、 N-溴代丁二酰亚胺(NBS,≥98%)、6-羟基萘醛(6-Hydroxy-2-naphthalde,≥97%)和四溴化碳(CBr4,分析纯)均购于百灵威科技有限公司;过氧化苯甲酰(BPO,≥97%)、三乙胺(≥99%)和CaH2(≥97%)均购自于百灵威科技有限公司);使用前,BPO溶于氯仿溶液中,以甲醇为沉淀剂反复精制2次后,真空干燥,保存备用;三乙胺经CaH2回流24 h,常压蒸出后,干燥密闭保存。 实验室用水为二次蒸馏水。
Varian Mercury plus 400M型核磁共振仪(NMR,美国瓦里安公司);Elemental Vario EL型元素分析(EA,德国艾力蒙塔);Finnigan-MAT ZAB-HS型气-质联用分析仪(MS,美国菲尼根玛特); TAInstruments SDT 2960型热重分析仪( TGA,美国TA仪器公司);Instruments Q100型差示扫描量热(DSC,美国TA仪器公司);Shimadzu Model 3100型紫外-可见光谱(UV-Vis,日本岛津公司);LS-55型荧光分光光度仪(美国Perkin Elmer公司);F97 Pro型荧光分光光度计(上海棱光技术有限公司);Leitz Laborlux 12型偏光显微镜(POM,德国徕卡),配有Leitz 350热台,可以程序升温和降温。
偏光显微镜(POM)实验在观察小分子单体的液晶偏光织构的时候,起偏片和检偏片是互相垂直正交的,以便将普通光改变为偏振光进行镜检,用以鉴别某一物质是否具有双折射性(各向异性)。 将适量小分子晶体样品直接置于洁净的圆形盖玻片(玻片事先用稀硝酸浸泡24 h、去离子水和丙酮清洗并干燥)上,在实际观察的时候,将另一片洁净的玻片盖在有小分子晶体的玻片上,小心放在热台上,为确定样品相变温度和充分观察其织构的变化,可调节升降温速率,或对样品施以一定的外力作用(例如按压或剪切)的方法。
电致发光器件采用旋涂成膜,真空热蒸镀阴极的方法制备。 滴2~3滴聚(3,4-乙烯二氧噻吩)-聚苯乙烯磺酸(PEDOT:PSS)在臭氧处理过的ITO玻璃基片上,在1000 r/min的转速下旋涂30 s。 旋涂完毕后,用去离子水擦除阴极和阳极上多余的PEDOT。 然后在120 ℃加热处理20 min后自然降至室温。 随后在PEDOT薄膜上将掺杂有质量分数8%液晶分子的4,4'-二(9-咔唑)联苯(cbp)溶液滴在发光区区域,在1000 r/min的转速下旋涂30 s。 旋涂完毕后,用二氯甲烷擦洗阴极,在65 ℃加热处理30 min后自然降至室温。 最后,使用蒸镀设备分别蒸镀1,3,5-三(1-苯基-1 H-苯并咪唑-2-基)苯(TPBi),LiF和Al层,作为注入电子的阴极。 器件的整体结构为:ITO/PEDOT:PSS 40 nm/CBP:X(旋)(40~60 nm)/TPBi 40 nm/LiF 1 nm/Al 100 nm。
1.4.1 5-溴-2-碘苄醇(1)的合成 在配有搅拌装置的1000 mL三口瓶中加入200 mmol (50.0 g)5-溴-2-碘甲苯,200 mmol (73.2 g) N-溴代丁二酰亚胺,5.2 mmol (1.2 g)BPO和约400 mL CCl4溶液,于70~80 ℃下反应。 反应7 h后,溶液颜色由无色转变为粉红色,瓶底有大量白色沉淀生成。 待反应冷却至室温后,过滤,滤除不溶物,滤饼用二氯甲烷洗涤数次,用TLC点板法检测直至滴出液无产物为止,收集滤液,除去溶剂,得到浅黄色固体。以石油醚为淋洗剂,柱分离提纯。得到白色固体5-溴-2-碘苄溴32.6 g,产率50%,mp 62~65 ℃。1H NMR(400 MHz,CDCl3), δ:4.52(s,2H,Ph—CH2),7.21~7.59(m,3H,Ar—H)。
按照参考文献[25]的方法,在配有搅拌装置,恒压漏斗的500 mL三口烧瓶中加入50.0 mmol(16.0 g)5-溴二碘苄溴, 60 mmol(4.9 g)乙酸钠,5 mmol(1.6 g)相转移催化剂四丁基溴化铵和300 mL水。 缓慢升温,回流20 h后,待反应冷却至室温,用30%NaOH调节溶液的pH值至11。 继续升高温度在110 ℃下回流反应24 h。 冷却后,有大量白色产物析出,过滤,将滤液用二氯甲烷萃取,旋干有机相溶液,柱分离提纯[ V(石油醚): V(乙酸乙酯)=3:1],得到白色固体产物5-溴-2-碘苄醇(化合物1)12.5 g,产率95%,mp 91~93 ℃。1H NMR(400 MHz,CDCl3), δ:2.03(s,1H,—OH),4.63(s,2H,Ph—CH2—),7.12~7.67(m,3H,Ar—);HRMS(EI,DIP): m/z计算值C7H6BrIO [M+H]+:314.86,实测值314.90。
1.4.2 对乙炔基苯胺(2)的合成 在配有搅拌装置和恒压漏斗的500 mL三口烧瓶中加入60.0 mmol(10.00 g)对溴苯胺、1.5 mmol(0.39 g)三苯基磷、0.9 mmol (0.66 g)双三苯基磷二氯化钯以及0.5 mmol(0.09 g)碘化亚铜,抽真空,通N2气,反复3次循环后加入约200 mL重蒸处理过的三乙胺,再用注射器将150 mmol(12.5 g)2-甲基-3-丁炔-2-醇加至反应体系中,慢慢升高温度,于80~90 ℃下,反应24 h。 待反应冷却至室温后,过滤,滤除不溶物,滤饼用无水乙醚洗涤数次,用TLC点板法检测直至滴出液无产物为止。 收集滤液,除去溶剂,得到深棕色粘稠液体。 柱分离提纯[ V(石油醚): V(EtOAc)=1:3],得到黄色固体对2-甲基-3-丁炔-2-醇炔胺7.1 g,产率70%,mp 112~116 ℃。1H NMR(400 MHz,CDCl3), δ:1.60(6H,—CH3),3.33(1H,—OH),6.57~6.63(2H,Ar),7.20~7.25(2H,Ar)。
在配有搅拌装置和恒压漏斗的500 mL三口烧瓶中加入0.06 mol(7.00 g)对2-甲基-3-炔-2-醇苯胺和0.18 mol(10.08 g)KOH,抽真空,通N2气,反复3次循环后加入约120 mL异丙醇,于80~90 ℃下回流反应12 h,溶液颜色逐渐由黄色转变为深棕色。 待反应完全冷却至室温后,反应液用硅胶过滤,用CH2Cl2反复冲洗,用TCL法检测直至滴出液无产物为止。 采用旋转蒸发仪旋干滤液后,柱分离提纯[ V(石油醚): V(EtOAc)=5:1],得到浅黄色固体产物对乙炔基苯胺(化合物2)5.3 g,产率75%,mp 99~103 ℃。1H NMR(400 MHz,CDCl3), δ:2.92(1H,—CH2),3.84(2H,—NH2),6.58~6.63(2H,Ar),7.28~7.32(2H,Ar)。
1.4.3 2-(4-乙炔苯胺基)-5-溴苄醇(3)的合成 在配有搅拌装置、回流装置的500 mL三口烧瓶中加入80.0 mmol 5-溴-碘苄醇(10.00 g)、6.8 mmol三苯基磷(1.78 g)、1.8 mmol双三苯基磷二氯化钯(1.26 g)以及3.4 mmol碘化亚铜(0.58 g),抽真空,通N2气反复3个循环后加入约300 mL重蒸处理的三乙胺,随后将溶有0.08 mol对乙炔基苯胺(9.36 g)的三苯胺溶液滴加至反应体系中,缓慢升高温度,回流反应24 h。 待反应冷却至室温后,过滤,滤除不溶物,滤饼用无水乙醚洗涤数次,用TLC点板法检测直至滴出液无产物为止。 收集滤液,采用旋转蒸发仪旋干滤液后得到棕黄色固体。 柱分离提纯[ V(石油醚): V(EtOAc)=2:1],得到约6.3 g黄色固体产物2-(4-乙炔苯胺基)-5-溴苄醇(化合物3),产率55%,mp 127~130 ℃。
1H NMR(400 MHz,DMSO-d6), δ:7.62(s,1H),7.41(dd, J=8.2,1.9 Hz,1H),7.32(d, J=8.2 Hz,1H),7.19(d, J=8.4 Hz,1H),6.55(d, J=8.5 Hz,1H),5.58(s,1H),5.42(t, J=5.7 Hz,1H),4.65(d, J=5.6 Hz,1H);HRMS(EI,DIP): m/z计算值C15H12BrNO [M+H]+:301.01,实测值301.04。
1.4.4 2-(4-乙炔苯胺基)-5-乙炔基苄醇(4)的合成 在配有搅拌装置、回流装置的500 mL三口烧瓶中加入7.97 mmol 2-(4-乙炔苯胺基)-5-溴苄醇(2.4 g)、0.93 mmol三苯基磷(0.24 g)、0.47 mmol双三苯基磷二氯化钯(0.33 g)以及0.69 mmol碘化亚铜(0.12 g),抽真空,通N2气,反复3次循环后加入约300 mL重蒸处理的溶剂三乙胺,用注射器将溶有7.97 mmol 2-甲基-3-丁炔-2-醇(0.67 g)的三苯胺溶液滴加至反应体系中,慢慢升高温度、回流反应24 h。 待反应冷却至室温后,过滤,滤除不溶物,滤饼用无水乙醚洗涤数次,用TLC点板法检测直至滴出液无产物为止。 收集滤液,采用旋转蒸发仪旋干滤液后得到深棕色粘稠液体。 柱分离提纯[ V(石油醚): V(EtOAc)=1:1],得到1.8 g黄色固体2-(4-乙炔苯胺基)-5-(2-甲基-3-丁炔-2-醇)苄醇,产率60%,mp 162~165 ℃。1H NMR(400 MHz,CDCl3), δ:7.51(s,2H),7.41(d, J=7.9 Hz,2H),7.32~7.26(dd, J=13.7,7.8 Hz,6H),6.64(d, J=7.6 Hz,4H),4.85(s,2H),3.86(s,2H),2.11(d, J=46.2 Hz,2H),1.62(s,6H)。
在配有搅拌装置的100 mL三口烧瓶中加入1.25 mmol 2-(4-乙炔苯胺基)-5-(2-甲基-3-丁炔-2-醇)苄醇(0.3810 g)和3.75 mmol KOH(0.2099 g),抽真空,通N2气,反复3次循环后加入约60 mL异丙醇,回流反应24 h后,溶液颜色逐渐由黄色转变为深棕色。 待反应完全冷却至室温后,反应液先用硅胶过滤,然后用CH2Cl2反复冲洗,用点板法检测直至滴出液无产物为止。 采用旋转蒸发仪旋干滤液后,柱分离提纯[ V(石油醚): V(EtOAc)=2:1],得到约0.25 g棕黄色固体产物2-(4-乙炔苯胺基)-5-乙炔基苄醇(化合物4),产率70%,mp 146~151 ℃。1H NMR(400 MHz,CDCl3), δ:7.55(d, J=7.8 Hz,2H),7.44(s,2H),7.40(s,1H),6.68(d, J=7.8 Hz,2H),5.45(s,2H),3.66(s,2H),3.14(s,1H);13C NMR(100 MHz,CDCl3), δ:153.15,144.34,138.76,135.68,132.02,128.99,124.81,121.41,119.28,115.15,97.83,83.51,78.04,74.19;HRMS(EI,DIP): m/z计算值C17H13NO [M+H]+:247.10,实测值247.15。
1.4.5 6-己氧基萘溴乙炔(5)的合成 在配有搅拌装置、回流装置的100 mL三口烧瓶中加入20.0 mmol(3.44 g)6-羟基萘醛,24.0 mmol(3.316 g)无水碳酸钾,抽真空,通N2气,反复3次循环后,用注射器加入24 mL溶剂乙腈,升高温度,在80~90 ℃下,反应0.5 h后,慢慢滴加溶于约2 mL乙腈中的20.0 mmol(3.302 g)溴己烷,滴加完后再回流反应16 h。 反应完后,冷却,抽滤,滤出碳酸钾等固体,滤饼用丙酮洗涤3次,收集滤液,采用旋转蒸发仪旋干滤液后,柱分离提纯[ V(石油醚): V(EtOAc)=5:1],得到5.1 g白色固体6-己氧基萘醛,产率90%,mp 81~83 ℃。1H NMR(400 MHz,CDCl3), δ:0.89~0.96(3H,—CH3),1.26~1.38(6H,—CH2—),1.80~1.90(2H,—CH2),4.08~4.15(2H,Ar—OCH2),7.17~7.22(2H,Ar),7.77~7.95(3H,Ar),8.26(1H,Ar),10.1(1H,Ar—CHO)。
在配有搅拌装置的250 mL三口瓶中加入50.0 mmol(14.50 g)三苯基磷,50.0 mmol(3.25 g)Zn粉,然后抽真空,通N2气,反复3次循环后,用注射器加入100 mL CH2Cl2并置于冰水浴中搅拌。 惰气保护下慢慢滴加溶有50.0 mmol(16.5 g)CBr4的CH2Cl2溶液。 待溶液滴加完后立即撤去冰水浴,在室温下继续反应24 h,直至溶液颜色由灰色转变为浅粉色,然后将溶有50.0 mmol(6.4 g)6-己氧基萘醛的CH2Cl2溶液慢慢滴入反应瓶中,溶液逐渐转变为灰色。 待反应完全后,过滤,滤出不溶物,滤饼用CH2Cl2/正己烷的混合溶剂反复冲洗,用TLC点板法检测直至滴出液无产物为止。 收集滤液,采用旋转蒸发仪旋干滤液后,柱分离提纯,以石油醚为淋洗剂,得到9.0 g乳白色固体6-己氧基萘乙烯二溴,产率88%,mp 90~93 ℃。1H NMR(400 MHz,CDCl3), δ:0.88~0.92(3H,—CH3),1.26~1.38(6H,—CH2—),1.81~1.88(2H,—CH2—),4.04~4.11(2H,Ar—OCH2—),7.11~7.18(2H,Ar),7.60(1H,Ar-CH=CBr2),7.63~7.76(3H,Ar),7.95(s,1H,Ar)。
在配有搅拌装置的500 mL三口瓶中加入12.0 mmol(5.00 g)6-己氧基萘乙烯二溴,13.0 mmol(1.50 g)叔丁醇钾,抽真空,通N2气,反复3次循环后用注射器加入约300 mL叔丁醇,在80~90 ℃下反应22 h。 待反应冷却至室温后,抽滤,滤出不溶物,滤饼用CH2Cl2洗涤数次,用TLC点板法检测直至滴出液无产物为止。 收集滤液,采用旋转蒸发仪旋干滤液后,柱分离提纯,以石油醚为淋洗剂,得到3.8 g黄色固体产物6-己氧基萘溴乙炔(5),产率95%,mp 106~110 ℃。1H NMR(400 MHz,CDCl3), δ:0.88~0.95(m,3H,—CH3),1.27~1.39(6H,—CH2—),1.78~1.88(2H,—CH2—),4.04~4.10(2H,Ar—OCH2—),7.08~7.19(2H,Ar),7.41~7.46(1H,Ar),7.62~7.70(2H,Ar),7.90(1H,Ar);13C NMR(100 MHz,CDCl3), δ:157.97,134.35,131.9,129.18,128.88,128.14,126.69,119.76,117.37,106.28,80.60,68.11,48.88,31.58,29.15,25.75,22.59,14.01。
1.4.6 5-{(6-己氧基萘基)丁二炔}-2-{(4-氨基苯基)乙炔}苄醇(6)的合成 在配备搅拌装置、回流装置的500 mL三口烧瓶中加入14.5 mmol 6-己氧基萘溴乙炔(4.8 g)、(0.477 g,1.82 mmol)三苯基磷、双三苯基磷二氯化钯(0.633 g,0.902 mmol)以及碘化亚铜(0.207 g,1.42 mmol),抽真空,通N2气,反复3次循环后加入约300 mL重蒸处理的溶剂三乙胺,随后用注射器将溶有2-(4-乙炔苯胺基)-5-乙炔基苄醇(3.0 g,12.2 mmol)的三苯胺溶液滴加至反应体系中,缓慢升高温度、回流反应24 h。 待反应冷却至室温后,抽滤,滤除不溶物,滤饼用无水乙醚洗涤数次,用TLC点板法检测直至滴出液无产物为止。 收集滤液,采用旋转蒸发仪旋干滤液后,柱分离提纯,得到深棕色粘稠液体。 柱分离提纯[ V(石油醚): V(CH2Cl2)]=1:1,得到约1.87 g黄色固体目标产物6,产率40%。 mp 125~130 ℃。1H NMR(400 MHz,CDCl3), δ:7.99(s,1H),7.68(dd, J=22.9,8.6 Hz,3H),7.56(d, J=8.4 Hz,1H),7.49(d, J=8.1 Hz,2H),7.46(d, J=11.3 Hz,2H),7.17(d, J=9.8 Hz,1H),7.09(s,1H),6.69(d, J=8.4 Hz,2H),5.48(s,2H),4.08(t, J=6.5 Hz,2H),1.88~1.80(m,2H),1.51(s,2H),1.37(s,4H),0.92(s,3H);13C NMR(100 MHz,DMSO-d6), δ:158.50,151.92,147.36,139.49,136.62,135.08,133.88,133.29,132.85,131.88,129.94,129.43,129.05,127.79,126.09,120.34,120.02,115.37,114.34,107.29,99.44,83.72,82.11,74.38,68.18,31.37,28.91,25.60,22.49,14.28;HRMS(EI,DIP): m/z计算值C35H31NO2[M+Na]+:519.24,实测值519.26。
化合物6属于热致型液晶,即温度的变化是发育出液晶相的条件,一般是随着温度的升高,液晶相态逐步发育出来。 那么在研究化合物的液晶行为之前,首先需要了解其热性质。
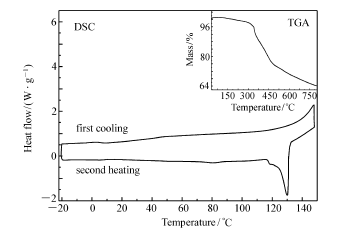
图1是化合物6的TGA曲线和DSC曲线。 由TGA图(图1插图)可知,化合物6被加热到300 ℃前仅发生了4%左右的热失重,这是由失水引起的。 此外,没有发生明显的热失重,说明其具有较好的稳定性。 而由DSC曲线可知,化合物6被加热到130 ℃的时候出现了一个明显的吸热峰,该峰对应的是晶体到液晶相的转变过程,随后在降温过程中没有观察到明显的从液晶相到晶体转变的放热峰,表明化合物6的晶体与液晶相之间转变的不可逆性。
 | 图1 化合物6二次升温和二次降温的DSC曲线。 插图:化合物6的TGA曲线Fig.1 DSC traces for compound 6 on second heating and subsequent cooling. Inset:the TGA traces of compound 6 from 50 to 800 ℃ |
{kind=link}
 | 图2 化合物6在135 ℃时的偏光显微镜图片Fig.2 Polarized light microscopy images of compound 6 at 135 ℃ A.the two black schlieren textures; B.the four black schlieren textures |
{kind=link}
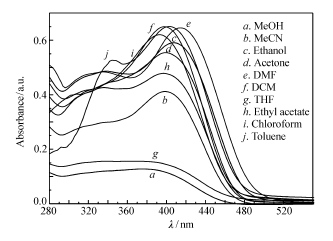
 | 图3 化合物6在不同极性溶剂中(浓度:2×10-6 mol/L)的紫外吸收光谱Fig.3 Absorption spectra of compound 6 with the concentration of 2×10-6 mol/L in different solvents |
{kind=link}
POM实验是最常用也是最直观的测量和判断液晶相态的手段,特别对于小分子液晶来说,通常情况下其形成的偏光织构多是具有特征性的典型织构,因此可以根据其偏光织构的特性来判断小分子形成的液晶相态。 化合物6在室温下为浅黄色针状晶体,将少量晶体散布在玻片上,从室温开始以10 ℃/min升温。可以观察到在125 ℃晶体开始融化,继续升温,到135 ℃时出现典型的纹影织构,即进入向列相液晶态。 织构见图2。
图3为化合物6在不同极性溶剂中(浓度:2×10-6 mol/L)的紫外吸收光谱,图中最大吸收峰位于400 nm附近。 从图可以看出,随着溶剂极性的增大,在400 nm附近紫外吸收峰波长变化不大,从极性较小的溶剂(乙醚)到极性较大的溶剂(甲醇),紫外吸收波长仅在1~18 nm之间移动。
图4为化合物6在不同极性溶剂中(浓度:2×10-6 mol/L)的荧光光谱。 荧光量子产率是荧光基团发出的光子数与吸收的光子数之间的比值,是考量荧光化合物光学性质的重要参数。 对于物质溶液的荧光量子产率的测定可以按照下式进行计算:
ϕ=ϕr(
式中, ϕ为荧光量子产率, A为荧光积分面积,OD为吸光度, n为折光指数,下标r代表已知荧光量子产率的参比物质。 实验中采用的荧光参比物质是硫酸奎宁,其室温下0.05 mol/L硫酸溶液的荧光量子产率为0.55±0.05。 通过测试及计算得到化合物6溶液在不同溶剂中的荧光量子产率。
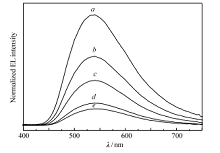
与紫外吸收光谱不同,化合物6的荧光发射光谱发生了显著的变化(如图4 A所示)。 由图4 A可以看出,随着溶剂极性从甲苯到DMF的增加,荧光发射光谱波长从486 nm红移至589 nm,同时,荧光量子效率从甲苯中的32%降低至1.6%。 在紫外灯照射下,化合物6在不同极性的溶剂中呈现出不同颜色的荧光。 例如,在低极性溶剂甲苯中发射蓝光,而在强极性溶剂DMF中发射橙光(如图4 B所示)。 这是由于化合物6分子中含有较长的炔键单元(-C≡C-),使共轭体系得以扩展,在光激发条件下,分子内存在着很强的电荷转移(ICT)能力。 在不同极性的溶剂中,这种ICT能力不同,因此产生发射峰的移动和发光颜色的不同[26]。 这种特性可用于经验性的判断不同有机溶剂的极性,为可视化识别提供了一种方法。
 | 图4 化合物6在不同极性溶剂中(浓度:2×10-6 mol/L)的荧光光谱( A);化合物6不同溶液在紫外光照下的照片(激发波长365 nm)( B)Fig.4 Fluorescence spectra of compound 6 in organic solvents of different polarity with the concentration of 2×10-6 mol/L( A), photographs of compound 6 in organic solvents of different polarity(at excitation wavelength of 365 nm( B) |
{kind=link}
我们又考察了材料的电致发光性能,由化合物6作为掺杂剂通过溶液成膜工艺制备了有机电致发光器件,结构为ITO/PEDOT:PSS(40 nm)/CBP:dopant( x%)(40~60 nm)/TPBi(40 nm)/LiF(1 nm)/Al(100 nm)。图5是器件在不同电压下的电致发光光谱,最大发射峰位于540 nm,发射黄绿光。 此外,在不同的电压下,发光光谱的峰形和峰位均没有太大的变化,表明其电致发光的稳定性较好。 器件的亮度-电压曲线和效率-电压曲线如图6所示,启动电压为7.20 V(亮度为1 cd/m2时),器件17.65 V时达到最大亮度126 cd/m2,最大流明效率为0.03l m/W,最大电流效率为0.21 cd/A。
 | 图5 化合物6制备器件在不同电压下的电致发光光谱Fig.5 Electroluminescence spectra of compound 6 at different voltages U/V: a.16; b.15; c.14; d.13; e.12 |
{kind=link}
 | 图6 化合物6制备的器件的亮度-电压曲线( A)和效率-电压曲线( B)Fig.6 Luminance( A), current efficiency and power efficiency( B) of compound 6 device versus voltage curves |
{kind=link}
本文通过多步Sonogashira偶联反应成功合成结构中含氨基的高共轭不对称芳炔有机化合物6,并对其液晶、溶剂化效应、光学性质及电致发光性能进行了研究。 结果表明,该化合物具有较高的晶体到液晶相的转变温度(135 ℃),形成向列相液晶。 将化合物6溶于不同极性的有机溶剂中,随着溶剂极性从甲苯到DMF的增加,其荧光发射光谱波长从在甲苯中的486 nm红移至DMF中的589 nm,并伴有荧光量子效率的降低。 在紫外灯照射下,化合物6在不同极性的溶剂中呈现出不同颜色的荧光,可用于经验型的判断不同有机溶剂的极性,为可视化识别提供了一种方法。 此外,采用旋涂的方式制备小分子电致发光器件,测试结果表明化合物6具有电致发光性能,启动电压为7.2 V,发绿光,是一种潜在的绿色电致发光材料。
本文属于开放获取期刊,遵循CCAL协议,使用请注明出处。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|