
SONG Kuanguang, LI Jian, GAO Chao, et al. Synthesis and Properties of Liquid Crystals with 3,3,3-Trifluoropropynyl Terminal Group[J]. Chinese Journal of Applied Chemistry, 34(6): 676-684


合成了16个三氟丙炔为端基的液晶化合物,以苯甲醛衍生物与1,1,1-三氯三氟乙烷的有机锌试剂进行加成脱水成烯,再通过Suzuki偶联反应后脱去HCl制得目标产物,总产率>67%,气相色谱(GC)纯度>99.5%,通过红外光谱(IR)、核磁共振谱(NMR)和质谱(MS)确定其结构。 利用DSC和POM对该类化合物的热性能进行了测试,结果表明,联苯类的化合物不具有液晶相,而三联苯类的化合物具有近晶A相,且熔点随烷基链碳原子的增加呈下降趋势。 物理性能测试结果表明,该类化合物的Δ ε值为12.61~21.94,末端三氟甲基基团和侧氟的引入可以有效的增加Δ ε值;Δ n值0.19~0.29,随分子内苯环数目的增加而变大; γ1值为49.9~468.0 mPa·s,随分子内苯环数量的增加而增加。 化合物4n具有高Δ ε(>19.2)、高 ε⊥(>10.3)及低Δ ε/ ε⊥(<1.9)。 通过模拟计算讨论了炔键位置对此类液晶材料性质的影响。 该类化合物在调节混合液晶介电各向异性和双折射率方面有很好的应用价值,同时通过分子结构的改进也可以很好地应用于FFS(fringe-field switching)显示模式。
Sixteen liquid crystals with 3,3,3-trifluoropropynyl terminal group were reported. The target compounds were synthesized via a series of reactions including the zinc reagent's reaction, Suzuki cross-coupling, and the elimination reaction of HCl. The total yields are more than 67%, and the gas chromatograph(GC) purities are more than 99.5%. The structures of the target compounds were characterized by infrared spectroscopy(IR), nuclear magnetic resonance(NMR) and mass spectrometry(MS). Moreover, their thermal properties were investigated by differential scanning calorimetry(DSC) and polarizing microscope(POM). The results show that the biphenyl type of compounds exists no liquid crystal phase while the terphenyl compounds have a smectic A phase. The melting points drop with the increase in the number of alkyl chain carbon. In terms of physical properties, these compounds have large Δ ε value(12.61~21.94), and the value of Δ ε is increased by trifluoromethyl and lateral fluorine substitution. The Δ n value is about 0.19~0.29 with the increase of the number of benzene rings. The γ1 value is in the range from 49.9 to 468.0 mPa·s, which goes up with the increase of the number of benzene rings. Compounds 4n have high Δ ε(>19.2), high ε⊥(>10.3), and low Δ ε/ε⊥(<2). The influence of the position of the acetylenic bond on the properties of this kind of liquid crystal was discussed by simulation. All of the above properties of these liquid crystals have been proved to be useful for tuning the birefringence and the dielectric anisotropy of the liquid crystal mixtures, and these liquid crystals also have good value for FFS(fringe-field switching) display mode by the improvement of molecular structure.


液晶显示已经成为现在主流的显示模式,拥有广阔的市场,液晶材料作为有机光电材料已经得到了长足的发展。 液晶材料往往需要具有大的介电各向异性(Δ ε,Δ ε=ε∥- ε⊥)以降低驱动电压而减小能耗,大的双折射率(Δ n,Δ n=ne- no)以降低液晶盒的盒厚而提高响应速度,低的旋转粘度( γ1)以进一步提高响应速度。 同时随着液晶显示模式的不断进步,液晶显示的画面质量也不断提高,但相比去传统的CRT显示器,液晶显示器的视角差是一大缺憾,而FFS(fringe-field switching)显示模式可以增大视角并提高透光率,克服了这一问题。 通常,负介电各向异性(Δ ε<0)液晶材料的透光率均高于正介电各向异性(Δ ε>0)液晶材料,但正介电各向异性液晶材料的驱动电压和响应速度均优于负介电各向异性材料。 研究表明,提高短轴方向介电常数( ε⊥)有利于提高正介电各向异性液晶材料的透光率[1]。 同时正介电各向异性液晶材料常常需要降低Δ ε/ε⊥来提高超扭曲显示电光特性曲线的陡度,从而提高多路驱动能力和对比度,达到改善视角关系,增大信息显示容量的效果[2,3,4],这也需要液晶材料具有大的 ε⊥值。 因此,具有高Δ ε、高 ε⊥、低Δ ε/ε⊥及大Δ n的液晶材料是FFS显示模式发展的一个关键点。
近年来,合成了多种侧链含有三氟烷基、三氟烯基的液晶化合物,其中若干种已得到良好的实际应用[5,6]。 通过比较,其中含有三氟丙烯基等不饱和基团的这类液晶化合物具有更大的介电各向异性值,适当的双折射率,更适用于液晶显示。 而三氟丙炔基团与三氟丙烯基团相比,能更进一步提高分子的π电子云密度,应具有更好的Δ n值[7],以及相近似或更高的Δ ε值。 目前已有少量该类液晶报导,例如:3,5,2'-三氟-4″-丙基-4-(3,3,3-三氟丙炔基)-[1,1',4',1″]三联苯(化合物4e,见表1),具有极大的介电各向异性值(Δ ε=31.6),较高的双折射率值(Δ n=0.277),较窄的液晶相区间:C 119.5 SA 125.3 I[5]。 此类液晶化合物具有较强的研究和应用价值。 但三氟丙炔基类化合物较多地用于医药化学品及农用化学品[8,9,10,11,12,13],较少应用于液晶材料方面。 该类液晶材料缺少系统性的研究,特别是在合成方法方面报道不多,导致性能规律认识不全面,基于此本文对三氟丙炔基类液晶材料的合成方法及性能进行了研究。 同时期望利用该类液晶Δ ε值很大的特点,设计出高Δ ε、高 ε⊥、低Δ ε/ε⊥及大Δ n的液晶单体。
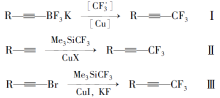
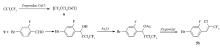
现有的三氟丙炔基类化合物合成方法主要有以下3种:1)由三氟硼酸钾类化合物反应生成三氟丙炔基类化合物[8,9,10],如Scheme 1式Ⅰ;2)由乙炔基类化合物反应生成三氟丙炔基类化合物[11,12,13],如Scheme 1式Ⅱ;3)由溴代乙炔基类化合物反应生成三氟丙炔基类化合物[5],如Scheme 1式Ⅲ。
 | Scheme 1 Synthetic methods of trifluoromethylated acetylenes |
以上的合成方法普遍存在原材料较贵,反应条件苛刻,收率较低的问题,而方法3)的收率仅为9.5%[5],且所给收率均为生成三氟丙炔基类化合物这一反应步骤的收率,前端化合物的制备也是一个较长的过程,会进一步影响成本和收率。
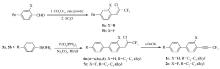
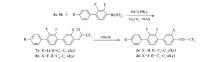
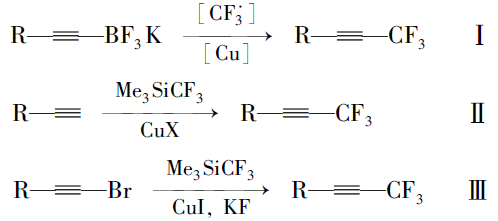
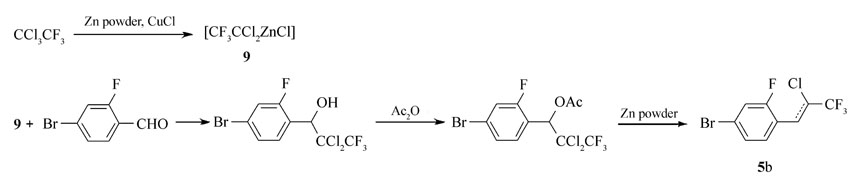
本文先由苯甲醛衍生物与1,1,1-三氯三氟乙烷反应制得中间体5a、5b,中间体5a、5b与多种芳基硼酸进行Suzuki偶联反应后再脱去氯化氢而制得目标产物。 该方法不再由炔烃出发制备产物,而是在分子一侧直接构建三氟丙炔基团,且改直线型的合成路线为偶联合成路线,更利于提高反应收率。 整体而言,该合成方法的原材料更便宜,反应条件温和,反应时间短,纯化简单,为该类液晶化合物合成提供了可行的新途径。 所合成的化合物1n(n=a,b,c,d)、2n、3n、4n的结构及合成路线如Scheme 2 和Scheme 3所示。
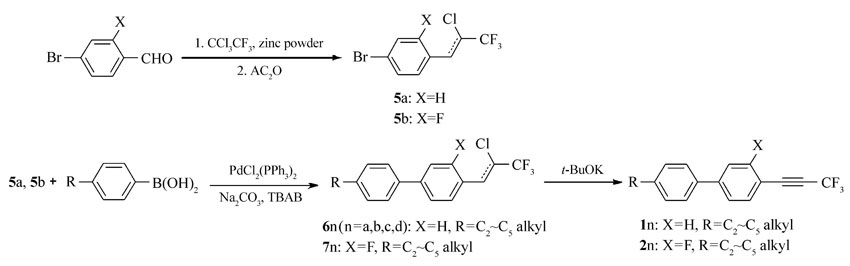 | Scheme 2 Synthesis route of compounds 1a~1d and 2a~2d |
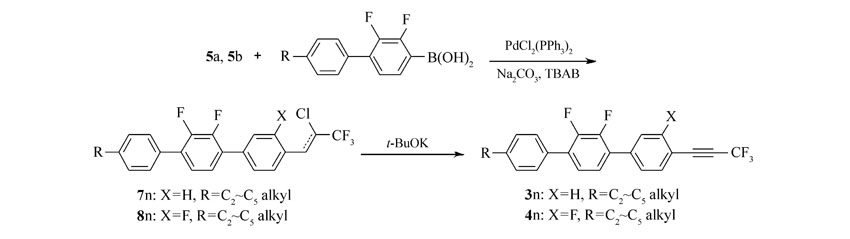 | Scheme 3 Synthesis of compounds 3a~3d and 4a~4d |
60SXR-FTIR型红外光谱仪(美国Nicolet公司);Bruke-AV 500MHz型核磁共振仪(NMR,德国布鲁克公司);7890B-7000C型气相色谱与质谱联用仪(GC-MS,美国Agilent公司);GC-2014C型气相色谱仪(日本岛津公司),载气为N2气,色谱柱为Restek Rxi-1 ms毛细管;DSC1型示差扫描量热仪(瑞士Mettler公司);6254型液晶性能测量系统(日本TOYO公司);EC-1型弹性常数测量系统(日本TOYO公司);NAR-4T型阿贝折光仪(日本ATAGO公司)。
1,1,1-三氯三氟乙烷、对溴苯甲醛、2-氟-4-溴苯甲醛、芳基类硼酸、双(三苯基膦)二氯化钯、四丁基溴化铵、叔丁醇钾、 N, N-二甲基甲酰胺、甲苯、乙醇为市售(成都市科龙化工试剂厂)工业级化工产品,锌粉、氯化亚铜、醋酸酐、无水碳酸钠为市售(成都市科龙化工试剂厂)分析纯化工产品。
1.2.1 4-溴-1-(2-氯-3,3,3-三氟-1-丙烯基)-2-氟苯(5b)的合成 参照文献[14,15]方法,1000 mL玻璃三口瓶,安装机械搅拌、顶端带干燥管的球形冷凝器及温度计。 向瓶中加入400 mL干燥DMF,40.95 g锌粉, 1,1,1-三氯三氟乙烷126.56 g,微量氯化亚铜。30 min后反应引发,迅速升温,滴加90.90 g 2-氟-4-溴苯甲醛的DMF溶液,滴加过程中温度迅速上升,加水浴冷却,反应保持50 ℃、1 h。 再加入73.44 g醋酸酐,58.50 g锌粉,迅速升温,水浴降温至50 ℃反应1.5 h。 将反应液倒入有1200 mL 10%稀盐酸的5 L烧杯中水解,充分搅拌后用150 mL×3正庚烷萃取有机相,并水洗至中性。 旋蒸除溶后得棕黄色液体(5b粗品),用正庚烷作为洗脱剂硅胶柱层析后得125.2 g无色透明液体(5b),具有顺反异构,异构体不影响下一步反应。顺反产物GC总含量:99.4%,产率92%。
1H NMR(500 MHz,CDCl3), δ:7.88(t, J=8.1 Hz, 1H),7.40(s,1H),7.36(d, J=8.5 Hz, 1H),7.31(d, J=10.7 Hz,1H);13C NMR(125 MHz,CDCl3), δ:119.37,119.57,122.38,124.71,124.79,127.60,127.63,130.53,159.31;IR(KBr), σ/cm-1:3075,1650,1600,1567,1483,1409,1291,1150,966,890,716,581,484;MS(70 eV) m/z(%):304.1(M+,100),269(11),187.1(28),169.1(23)。
1.2.2 4-(2-氯-3,3,3-三氟丙-1-烯基)-2',3,3'-三氟-4'-丙基-1,1':4',1″-三联苯(8b)的合成 N2气保护下,向单口瓶中加入15.2 g产物5b(溶于50 mL甲苯),15.19 g丙基苯基-2,3-二氟苯硼酸(溶于50 mL乙醇),15.9 g无水碳酸钠(溶于50 mL水),1.61 g四丁基溴化铵,0.3505 g双(三苯基膦)二氯化钯。 加热回流反应6 h。 降至室温,用50 mL×3正庚烷萃取反应液,有机相水洗至中性,旋蒸除溶得淡黄色固体(7b粗品),用正庚烷作为洗脱剂硅胶柱层析后得17.52 g白色固体(7b),具有顺反异构,异构体不影响下一步反应。 顺反产物GC总含量:98%,产率77%。
1H NMR(500 MHz,CDCl3), δ:8.10(t, J=7.9 Hz,1H),7.52(s,1H),7.48(d, J=7.8 Hz,2H),7.43(d, J=8.3 Hz,1H),7.36(d, J=11.2 Hz,1H),7.30~7.19(m,4H),2.64(t, J=7.6 Hz,2H),1.77~1.60(m,2H),0.98(t, J=7.3 Hz,3H);13C NMR(125 MHz,CDCl3), δ:13.86,24.47,37.84,115.86,116.05,119.27,119.37,122.79,124.22,124.46,124.98,127.08,127.15,128.70,128.72,128.86,129.80,131.19,131.56,138.56,143.31,147.64,149.65,161.70;IR(KBr), σ/cm-1:2963,1913,1620,1564,1405,1288,1148,964,808,659,507;MS(70 eV) m/z(%):454.1(M+,55),425.1(100),212.7 (7)。
1.2.3 2',3,3'-三氟-4″-丙基-4-(3,3,3-三氟丙-1-炔-1-基)-1,1',4',1″-三联苯(4b)的合成 向单口瓶中加入9.08 g产物7b,50 mL甲苯,搅拌溶解后加入2.91 g叔丁醇钾,室温反应1 h。 用50 mL×3正庚烷萃取反应液,有机相水洗至中性,旋蒸除溶得目标微黄色固体(4b粗品),用正庚烷作为洗脱剂硅胶柱层析后得8.02 g白色固体(4b),GC:99.6%,产率96%。
1H NMR(500 MHz,CDCl3), δ:7.60(t, J=7.5 Hz,1H),7.50(s,1H),7.48(s,1H),7.39(t, J=10.2 Hz,2H),7.28(t, J=6.4 Hz,3H),7.25~7.21(m,1H),2.69~2.60(m,2H),1.75~1.63(m,2H),1.04~0.94(m,3H);13C NMR(125 MHz,CDCl3), δ:13.86,24.46,37.81,80.05,80.10,106.86,106.98,115.69,124.21,124.73,125.08,126.72,126.77,128.69,128.71,128.87,131.43,131.47,131.54,134.11,143.41,147.50,147.62,162.36;IR(KBr), σ/cm-1:2960,2250,1620,1480,1460,1310,1160,814,681,662,525;MS(70 eV) m/z(%):418.2(M+,60),399.1(5),389.2(100),299.1(6)。
其它化合物的表征见辅助材料。
本文的合成方法主要有3步,首先利用有机锌试剂构建端基,然后通过Suzuki偶联反应构建分子主体结构,最后脱去端基上的HCl从而得到目标产物。 三步反应条件均较温和,收率高,也易于操作。 其中端基的构建尤为重要,也是本文合成方法的难点。 在文献中[14],中间体5的合成采用的是一锅法,将锌粉,1,1,1-三氯三氟乙烷,氯化亚铜,苯甲醛衍生物同时加入反应瓶中,再等待反应引发。 但实际操作时,发现原料为对溴苯甲醛时,反应的引发时间大于5 h;原料是2-氟-4-溴苯甲醛时,反应无法引发。 经查阅文献[15]发现,该反应的引发为两步,如Scheme 4 ,先由锌粉与1,1,1-三氯三氟乙烷反应生成锌的有机试剂,然后与醛类化合物进行亲核反应。 所以本方法先制备有机锌试剂,再加入醛类化合物反应,反应时间大大缩短,仅为3 h。
 | Scheme 4 Synthesis mechanism of compound 5b |
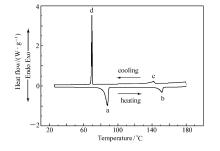
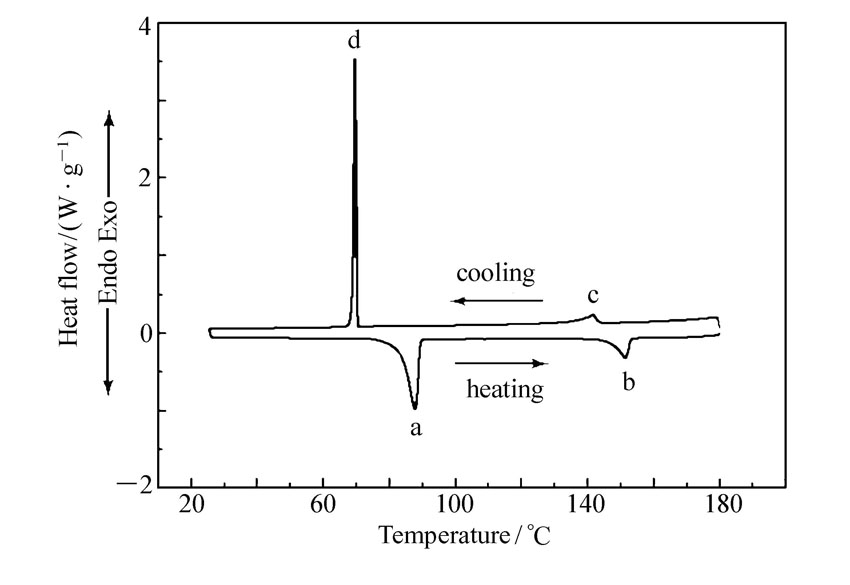 | 图1 化合物4c的DSC曲线Fig.1 DSC trace of compound 4c |
 | 图2 化合物4c的近晶A相(125 ℃)偏光显微镜照片Fig.2 SA phase photomicrograph of compound 4c at 125 ℃ |
在N2气氛围下,利用DSC以5 ℃/min的升降温速率对所合成化合物的相变温度进行测量(图1为化合物4c的DSC曲线),并结合偏光显微热分析仪(POM)进行了相态确认(图2为化合物4c的POM照片),结果如表1所示。图1中曲线的下半部分为升温过程,上半部分为降温过程,峰a为化合物4c晶体向液晶态转变的吸热峰,峰b为液晶态向液态转变的吸热峰,即清亮点,峰a至峰b区间即为升温过程的液晶相区间;峰c为液态向液晶态转变的放热峰,峰d为液晶态向晶体转变的放热峰,峰c至峰d区间即为降温过程的液晶相区间。 结合DSC曲线,经偏光显微热分析仪观察,化合物4c在液晶相区间呈近晶A相。
| 表1 目标化合物的相变温度 Table 1 Phase transition temperature of the target compounds |

从表1可以看出目标产物中,联苯类的产物没有液晶相,三联苯类的产物为互变液晶,属近晶A相;在末端苯环上的侧向氟取代使产物的熔点下降,有利于提高与其它化合物的相溶性。 化合物的液晶区间随着烷基链的增加而变宽,化合物3b、4b与化合物4e相比,液晶区间得到了较大的拓展,这是因为化合物4e的分子两侧均有氟取代导致了分子的长径比减小,从而限制了液晶区间,而化合物3b、4b只有分子一侧有氟取代,保证了较大的长径比。 从图3可以看出,随着端基烷基链碳原子的增长,化合物的熔点均呈下降趋势;化合物4n的清亮点具有明显的奇偶效应,即烷基链为奇数碳的化合物的清亮点高于偶数碳的化合物。
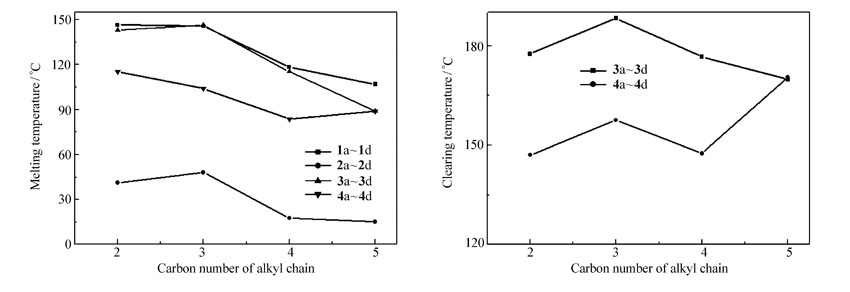 | 图3 烷基链碳原子数对目标产物熔点和清亮点的影响Fig.3 Effect of alkyl chain carbon number on melting temperature and clearing temperature of target compounds |
所测化合物与基础配方(Host)以5∶95质量分数比例进行混合(其中化合物2a为15∶85),加热至清亮点后混合均匀。采用阿贝折光仪(25 ℃,589 nm)测试混合液晶的Δ n;采用弹性常数测量系统测量Δ ε、 ε⊥;采用液晶性能测量系统测量 γ1。 Δ ε、 ε⊥、Δ n近似满足加法规则,可以外推出化合物单体的Δ ε、 ε⊥、Δ n值, γ1使用公式ln γm= x·ln γh+(1- x)ln γc进行外推, γm为混合后的旋转粘度, γh为基础配方的旋转粘度, γc为目标产物的旋转粘度, x为基础配方的质量分数。 所选用的基础配方(Host)由反式-4-乙基双环己基-3,4-二氟苯、反式-4-丙基双环己基-3,4-二氟苯、反式-4-戊基双环己基-3,4-二氟苯三种液晶单体化合物按照质量比1∶1∶1配成。 测试结果如表2所示。
| 表2 化合物的物理性能测试数据 Table 2 Physical properties of target compounds |
表2数据看出,随着柔性烷基链的碳原子数目的增加,Δ ε值呈下降趋势,但目标化合物的Δ ε值均大于12.6,最高达21.05,同时在基础配方中加入目标产物后,介电各向异性值Δ ε具有较大幅度上升,这对提高混晶配方的Δ ε值具有明显的作用。 主要原因是分子末端的—CF3基团具有很强的吸电子能力,可使分子长轴方向的偶极矩得到很大的增加,从而促使目标产物整体具有较高的Δ ε值。 化合物2n与1n、4n与3n相比,Δ ε值均有大幅提升,化合物4n甚至超过化合物3n 50%,这是因为分子末端侧向引入了氟原子,大幅度增加分子长轴方向的偶极矩,从而增加Δ ε值。
从表中可以看出,化合物2n、3n和4n均具有高的 ε⊥(>6.12),这是因为这3个系列化合物均具有侧向氟取代,提供了短轴方向的偶极矩,从而增大了 ε⊥。 由文献[16]可知,苯环2,3位的极性取代基可以为短轴方向提供偶极矩分量,而长轴方向偶极矩分量会相互抵消,从化合物3b、4b的Δ ε比化合物4e有所降低可以看出2位的氟取代可以增加 ε⊥,但不利于提高Δ ε。 故在设计分子时,化合物4n系列比化合物3n系列在分子末端的苯环上增加了一个侧氟取代,这样可以在提供短轴方向偶极矩分量的同时也提供长轴方向的偶极矩分量,从测试结果也可看出,化合物4n的Δ ε较化合物3n有了近53%的提升, ε⊥有了近50%的提升。 当液晶材料的Δ ε/ε⊥<2时,可以达到较好的显示效果[3,4],而化合物3n及化合物4n的Δ ε/ε⊥均小于1.9。
从表2可以看出,该类液晶均具有大的双折射率Δ n值,且随着柔性烷基链的碳原子数目的增加Δ n值也呈微弱下降趋势。 化合物的Δ n值均高于0.19,最高达0.2866。 这是由于分子中炔键的引入,提高了分子的线性共轭程度,使分子的π电子云密度得到很大提高,促进分子的Δ n值提升。 化合物2n与1n、化合物4n与3n相比,Δ n值均有一定程度的降低,这可能是因为侧向氟原子的引入所导致。 氟原子具有较强的吸电子能力,使电子云向侧向氟原子方向移动,降低了长轴方向的π电子云密度,导致了Δ n值的降低。 随着柔性烷基链的碳原子数目的增加,Δ n值也呈下降趋势。 化合物3b、4b与4e有近似的π电子云密度,故Δ n值相近。
从表2可以看出,联苯体系具有非常低的旋转粘度 γ1,但随着苯环的增加, γ1值迅速增加,这可能是因为三联苯体系分子的体积更大,刚性更强,分子的排列也更加有序所导致。 而从表中可以明显看出末端侧向氟原子的引入,可以一定程度降低产物的 γ1值。 化合物1n及2n系列化合物具有极低的旋转粘度( γ1<55 mPa·s)。
化合物4e分子末端苯环上的两个侧位氟原子可以增加长轴方向的偶极矩,但分子短轴方向的偶极矩分量会相互抵消,不利于 ε⊥值的提高,而化合物4b在化合物4e分子结构的基础上将末端苯环上的一个侧氟移至第二个苯环的侧位上,这样Δ ε值虽然有所降低,但仍达到了19.29的高值,并且 ε⊥高达10.38。 经过结构调整的化合物4n系列化合物具有高Δ ε、高 ε⊥、低Δ ε/ε⊥及大Δ n。
为了进一步研究炔键在不同位置对化合物液晶性能的影响,合成了炔键作为中心桥键的类似化合物1e、1f,并与化合物1a、1d进行对比。 利用Vuks式(1)、(2)、Maier and Meier式(3)[17]及半经验量子化学(AM1)模拟的方法进行了计算。 Vuks公式和Maier and Meier公式将化合物宏观的物理性质(折射率、介电常数)与分子的微观属性(极化率、偶极矩)相联系起来。
在这里Δ n=ne- no, n2=(
分子的
| 表3 化合物 |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
从表3可以看出,化合物1a与1e、1d与1f有相近
合成了16种三氟丙炔基类液晶化合物,并对其液晶性能和物理性进行了研究。 中间体5a、5b的合成,使分子的构建更加的简便,可合成的液晶化合物结构多样性增加,有利于设计合成性能更好的新型液晶材料。 而三氟丙炔基类液晶化合物分子中三氟甲基的引入可以有效的增加分子的Δ ε值,端基炔键的引入可以大幅提升Δ n值,末端侧向氟原子的引入,可以增加分子的Δ ε值。 与炔键作为中心桥键的类似化合物相比,三氟丙炔类单体液晶具有更好的液晶性能。 化合物1n、2n系列化合物具有高Δ ε、大Δ n及极低的旋转粘度,适用于调节混合液晶的双折射率、介电各向异性及旋转粘度。 而化合物4n系列化合物具有高Δ ε、高 ε⊥、低Δ ε/ε⊥及大Δ n,可用来提高混合液晶的 ε⊥及降低Δ ε/ε⊥,并应用于FFS显示模式。 以上研究表明,三氟丙炔类液晶有着十分巨大的潜在研究及应用价值。
辅助材料(Supporting Information)[目标产物的结构表征数据]可以免费从本刊网站(http://yyhx.ciac.jl.cn/)下载。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|