
WU Qiang, KANG Chuanqing, GAO Lianxun. Advances in Olefin Isomerization Reactions Catalyzed by the First-row Transition Metals[J]. Chinese Journal of Applied Chemistry, 34(1): 25-39


过渡金属催化的烯烃异构反应在有机化合物合成、日用化学品合成、原料油应用和天然产物合成中都有着举足轻重的作用。 廉价过渡金属由于其在资源、价格、后处理等方面的优势,日益受到研究者的重视。 本文主要综述了近几十年来廉价过渡金属铁、钴、镍在催化烯烃异构反应方面的研究进展,详细阐述了不同的催化体系在催化活性、反应选择性、底物适用性及反应机理方面的特点。 虽然目前的催化体系已经表现出优异的性能与应用价值,但在烯烃异构的立体选择性、区域选择性机制等方面,仍然需要更深入的研究。
Transition-metal-catalyzed olefin isomerization has played a crucial role in the synthesis of organic compounds, commodity chemicals, natural products, and petroleum feedstocks. Although noble metals such as Ru, Rh and Ir as catalysts for the conversions have made dramatic achievements, the disadvantages of the rare resource, high cost, and poisoning products have limited its wide application in the synthesis. Due to the abundant supply, low cost and easy removal from the final products, the first-row transition metals have attracted much attention from the chemists and have proven to be very promising with respect to the development of more sustainable catalytic system in olefin isomerizaton. This review summaried the advances in olefin isomerization catalyzed by iron, cobalt, nickel in the past years and illustrates the differences among different catalytic systems in catalytic activity, regio- and stereo-selectivity, and substrate scope. Recent developments in the olefin isomerization catalyzed by the first-row metals have proven to be very promising and effective. The reaction mechanism was discussed to have deep insight into the isomerization. The mechanism of olefin isomerization was illustrated clearly in the reported articles through intermolecular or intramolecular hydrogen shift, and the stereoselectivity and regioselectivity in different catalytic system was still worthy of further investigations. Further studies in the field to solve the challenges in high catalytic activity, stereoselectivity, and wide substrate scopes would bring the isomerization into better prospect.

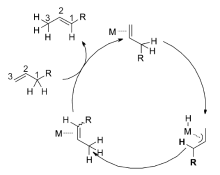




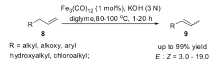


烯烃作为有机化合物基本骨架,大量存在于石油化工原料及产品、天然产物和合成分子中。 由于碳碳双双键的存在,它也可以作为一类功能化合物,通过对双键的修饰获得新的化合物,比如烯烃的氢甲酰化[1]、硼氢化[2,3,4]、氢硅烷化等[5,6]。 但天然存在的烯烃种类很少,为了满足不同反应的需求,各种烯烃的合成方法被开发出来,如消除反应、偶联反应、脱氢反应、烯烃复分解反应等。 通过对已有双键的异构反应构建新的烯烃也是一种高效的烯烃合成方法,烯烃的异构不会破坏原有分子的基本骨架,只是双键发生迁移,生成新的烯烃,不会产生大量的副产物,没有新的碳碳键的生成,是一种原子经济性的反应。 烯烃的异构,一种是在碱的作用下来实现双键的迁移,另外一种是通过过渡金属催化来实现。 相对于碱催化的反应,过渡金属催化的烯烃异构由于其高效性、高选择性获得了更多的关注。
过渡金属催化的烯烃异构是指烯烃1在金属催化剂的参与下,双键沿着碳链发生迁移,生成具有一定顺反( Z/E)比例的混合2-烯烃,在活性催化物种[M]的作用下,双键会继续发生迁移生成各种烯烃的混合物。 在不同的金属催化体系下,烯烃的异构会得到不同的产物(Scheme 1)[7]。
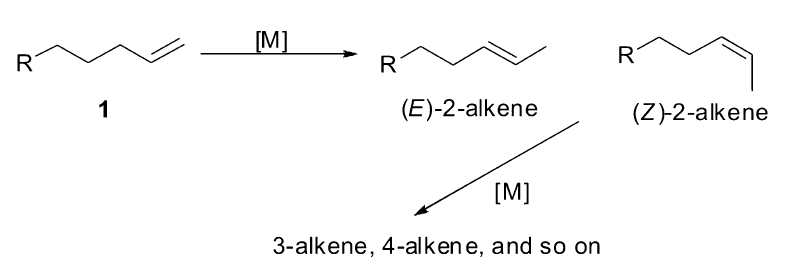 | Scheme 1 Transition-metal catalyzed olefin isomerization[7] |
{kind=link}
过渡金属催化的烯烃异构反应在有机化合物合成、日用化学品合成、原料油应用和天然产物合成中均有着举足轻重的作用[8,9,10,11,12]。 上世纪七八十年代起,高效的过渡金属催化的烯烃异构反应开始受到各国研究者的关注。 从针对特定化合物合成的催化体系到普遍适用的催化体系,从价格昂贵的贵金属(Pd、Rh、Ru、Ir等),到价格低廉的过渡金属(Fe、Co、Ni等),各种催化体系被开发出来应用于烯烃的异构反应,取得了令人瞩目的成就。 事实上,使用廉价过渡金属替代贵金属实现过渡金属催化的反应,代表了面向更加环境友好的合成化学发展方向,一直是研究领域广泛追求的目标[13,14]。 虽然在催化烯烃异构方面,贵金属Pd[15,16]、Rh[17,18,19]、Ru[20,21,22,23,24,25]、Ir[26,27,28]等表现出优异的催化活性与选择性,但随着现代工业科技的发展,贵金属由于自身资源匮乏、价格昂贵、毒性大、后处理繁琐,加上产品中对其残余量的严格标准导致贵金属的应用受到很大的限制。 廉价过渡金属虽然催化活性上稍逊一筹,但是在价格、资源、后处理等方面的优势使得其具有更大的应用前景。 基于此,为了更深入了解廉价过渡金属催化的烯烃异构反应,本文将回顾近几十年来廉价过渡金属在催化烯烃异构反应方面的研究进展。 由于对于廉价过渡金属的研究主要集中在金属铁、钴和镍上,所以本文主要总结了这三种金属在催化烯烃异构反应方面的发展现状。
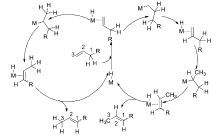
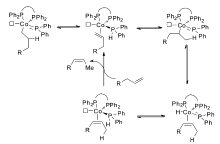
经过多年的研究发现,过渡金属催化的烯烃异构反应机理主要以两种方式进行:1)通过过渡金属与烯丙基形成复合物中间体( π-allyl)来诱导双键的迁移[23,26,27,29,30],见Scheme 2;2)金属氢键(Met—H)对双键的1,2-加成消除机理[7,31,32,33,34],见Scheme 3。 这两种反应机理虽然都是通过氢迁移来实现双键的迁移,但具有本质上的区别。 对于 π-allyl反应机理,首先是过渡金属与双键配位之后,通过氧化加成生成 π-allyl中间体来促进双键的迁移,发生分子内的1,3-氢迁移,而1,2-加成消除机理首先是原位生成的活性物种Met—H与双键配位,对双键的马氏与反马氏加成得到烷基金属复合物(M-alkyl)之后,通过 β-氢消除来达到双键迁移的目的,发生分子间的1,2-和1,3-氢迁移。
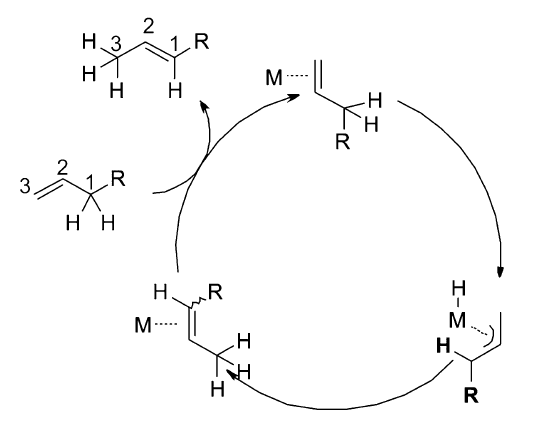 | Scheme 2 π-Allyl mechanism |
{kind=link}
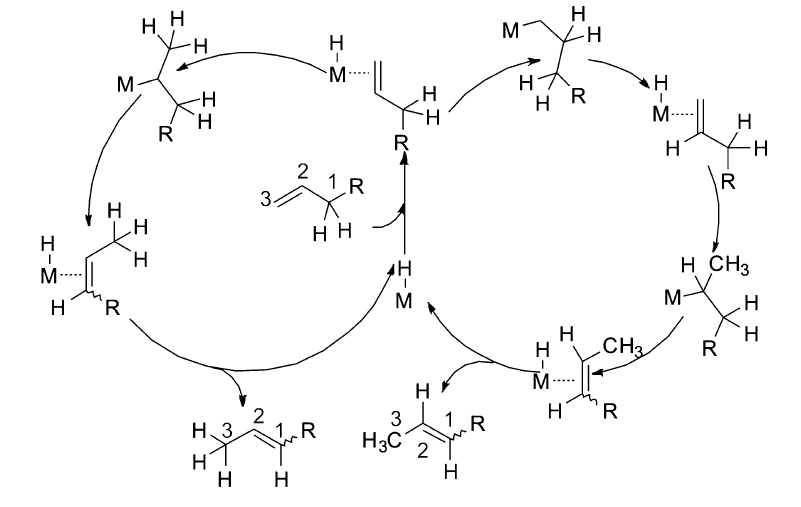 | Scheme 3 Metal-hydride insertion-elimination mechanism |
{kind=link}
金属铁催化剂由于其相对低廉的价格、较好的反应活性获得较多的关注。 近些年的研究发现,铁作为催化剂在偶联反应、氧化反应、氢化反应等[35,36,37]其它一些反应中表现出较高的活性,对于其在烯烃异构反应的研究很早就出现了。 早在1966年,Davison小组[38]研究发现,化学计量的多羰基铁复合物Fe(CO)5可以促进不饱和的脂肪酯异构得到共轭的脂肪酯。 在这之后的很长一段时间里,关于铁催化的烯烃异构都是使用多羰基铁复合物作为催化剂。 1973年,Casey小组[39]使用催化剂量的多羰基铁复合物Fe3(CO)12作为催化剂,催化3-乙基-1-戊烯(2)的异构反应。 在该催化体系下,异构是以分子内的1,3-氢迁移的机理进行的,最终生成热力学稳定的产物3-乙基-2-戊烯(3)(Scheme 4)。
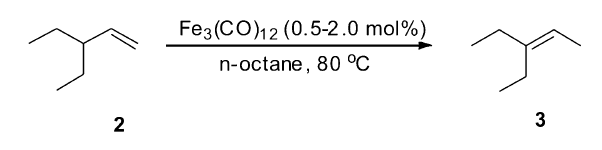 | Scheme 4 Fe3(CO)12 catalyzed isomerization of 3-ethyl-1-pentene[39] |
{kind=link}
早期的铁催化烯烃异构主要针对基础与机理研究,直到1977年,De Pasquale[40]首次将多羰基铁复合物催化体系成功运用至茴香醚(5)的合成中。 使用催化剂量的Fe(CO)5,在无溶剂体系下,最多可将45 g艾草醚(4)高效转化为茴香醚(5)(Scheme 5)。 虽然反应需要在高温条件下进行,但这为烯烃异构催化体系提供了实际的应用前景。
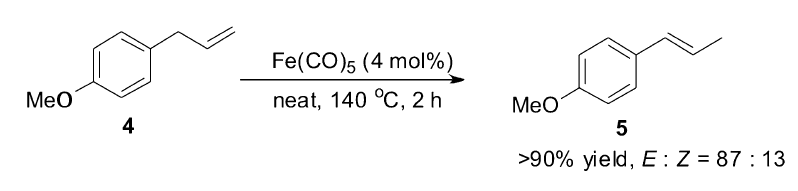 | Scheme 5 Fe(CO)5catalyzed isomerization of estrogole[40] |
{kind=link}
1985年,Darensbourg小组[41]发现,含铁的杂双金属催化剂[HFeM(CO)8L-](M=Cr,Mo,W;L=CO,PR3)在光催化作用下,温和条件下可以高效地将烯丙基苯(6)异构转化为1-丙烯基苯(7)(Scheme 6)。 简单的杂双金属氢化合物[HFeM(CO
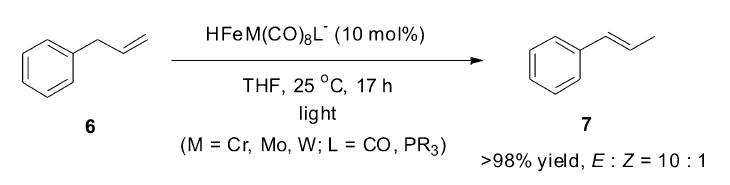 | Scheme 6 [HFeM(CO)8L-] catalyzed isomerization of allylbenzene[41] |
{kind=link}
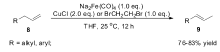
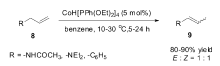
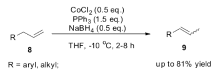
1995年,Periasamy小组[42]研究发现Fe(CO)5复合物在萘钠还原下形成的化合物Na2Fe(CO)4,在氯化亚铜或1,2-二溴乙烷作为氧化剂的作用下,可以有效将端烯烃(8)转化为2-烯烃(9)(Scheme 7)。 当Na2Fe(CO)4与CuCl作为反应体系时,端烯烃(8)可以完全转化为 E式2-烯烃(9),而不是 E/Z混合的2-烯烃,但Na2Fe(CO)4与BrCH2CH2Br作为反应体系时,生成的异构产物为 E/Z混合的2-烯烃。 虽然该反应体系具有非常高的立体选择性与区域选择性,但Na2Fe(CO)4必须化学计量使用。
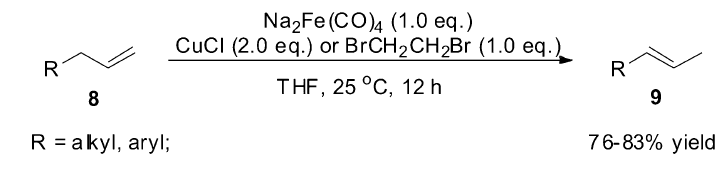 | Scheme 7 Na2Fe(CO)4 catalyzed olefin isomerization[42] |
{kind=link}
1998年,Kong小组[43]发现,催化量的Fe(CO)5复合物与NaOH原位生成的NaHFe(CO)4在非常温和的条件下高效地催化烯丙基醚类衍生物(10~12)异构生成烯基醚类化合物(13,14)(Scheme 8)。 该催化体系不仅适用于各类烯丙基醚类衍生物,也可以催化烯丙基苯类化合物的异构反应,生成高选择性的 E式烯烃。 虽然该催化体系已经具有相当高的活性及应用价值,但是该催化体系是否适用于更多类型烯烃的异构反应,至今未见报道。
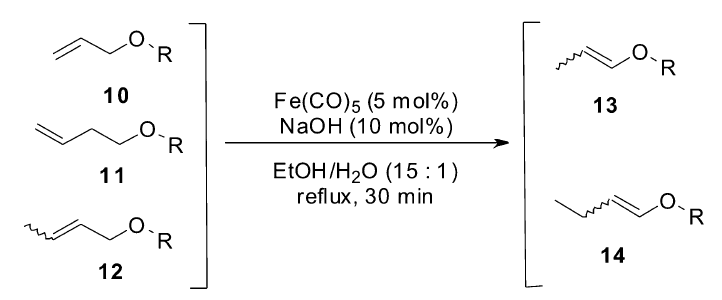 | Scheme 8 Fe(CO)5/NaOH catalyzed isomerization of allyl ethers[43] |
{kind=link}
除了Fe(CO)5复合物具有催化烯烃异构的能力,其它类型的多羰基铁复合物同样也是可以作为催化剂类来催化烯烃的异构反应。 2012年,Beller小组[44]发现,相对于其它的多羰基铁复合物,十二羰基三铁复合物Fe3(CO)12作为催化剂具有最高的催化活性,可以高选择性将端烯烃异构为2-烯烃,即使延长反应时间2-烯烃也不会继续发生异构生成烯烃混合物。 理论模拟计算(DFT)和扩展X射线精细结构(EXAFS)解析表明,反应中真正的催化活性中间体是[HFe3(CO
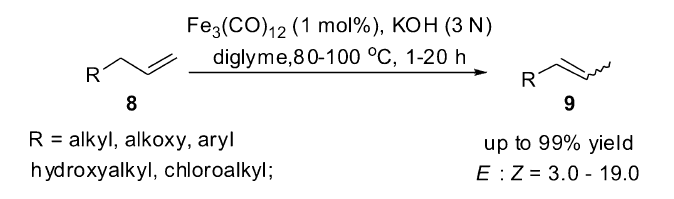 | Scheme 9 Fe3(CO)12/KOH catalyzed olefin isomerization[44] |
{kind=link}
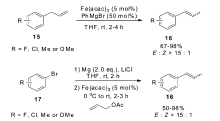
除了多羰基铁复合物作为催化剂,其它类型的铁催化剂也具有催化烯烃异构的能力。 2011年,Wangelin小组[45]发现,以Fe(acac)3作为催化剂,以PhMgBr作为还原剂,可在室温条件下快速催化烯烃的异构反应。 该催化体系不仅高效催化大量烯丙基苯类化合物15的异构,并且控制异构产物的 E:Z比大于15:1,也可以有效催化直链烯烃的异构,控制区域选择性的同时,也可以很好地得到较高的 E/Z比。 为了提高该催化体系的适用范围,该小组开发出一锅法多米诺反应,由溴代芳烃17出发,先发生铁催化的烯丙基化反应,之后再发生铁催化的烯烃异构反应,这为1-丙烯基苯类化合物16的合成提供了一条更环保的合成方法(Scheme 10)。 通过氘代底物的实验表明,在该催化体系下,烯烃的异构是以分子内的1,3-氢迁移机理进行。
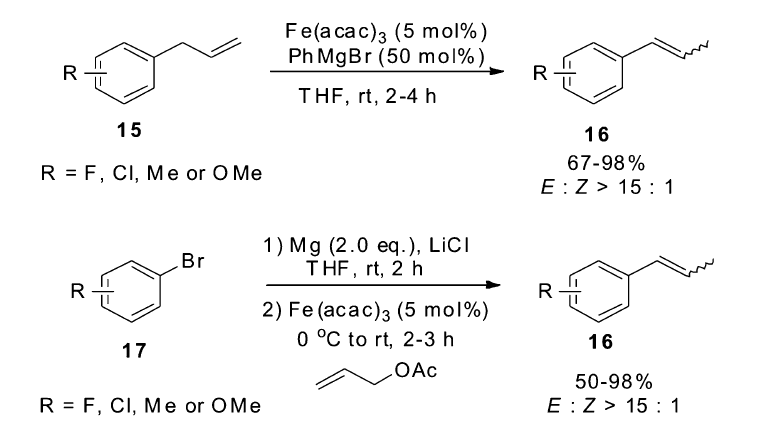 | Scheme 10 Fe(acac)3/PhMgBr catalyzed isomerization of allylbenzenes[45] |
{kind=link}
经过多年的发展,在铁催化烯烃异构方面已建立起多个催化体系,但大多缺乏系统性研究,最引人注目的还是近年来Beller小组[44]开发的Fe3(CO)12/KOH体系和Wangeli等[45]的Fe(acac)3/PhMgBr体系,这两个体系不仅具有高效的反应活性,还可以广泛适用于各种类型的烯烃,具有很高的应用价值。
金属钴在催化烯烃异构反应方面同样也显现出高效的催化活性,后期发展过程中发现,钴催化体系能够表现出与其它过渡金属完全相反的立体选择性。 1961年,Orchin小组[46]在研究以HCo(CO)4作为反应物的1-戊烯甲酰化反应的过程中发现,反应结束后,剩余的过量1-戊烯几乎全部转化为2-戊烯( E式为主),说明在该反应体系下HCo(CO)4促成了烯烃的异构反应。 起初,该小组分别以HCo(CO)4与DCo(CO
1986年,Onishi小组[49]研究发现氢化钴复合物CoH[PPh(OEt)2]4在光辅助的作用下生成的CoH[PPh(OEt)2]3,能够高效催化烯丙基衍生物(8)的异构反应(Scheme 11),但是对于烯丙基醚类的底物,在同样的催化体系下却会发生C—O键的断裂生成丙烯。 他们认为,在这种光辅助的条件下,产生的非饱和的钴复合物促进了异构反应的发生,该小组的后续研究发现,CoH[PPh(OEt)2]4复合物[50]在电化学氧化下生成的一种17电子的Co(Ⅱ)复合物确实可以在非光照条件下很好地催化烯烃的异构反应。
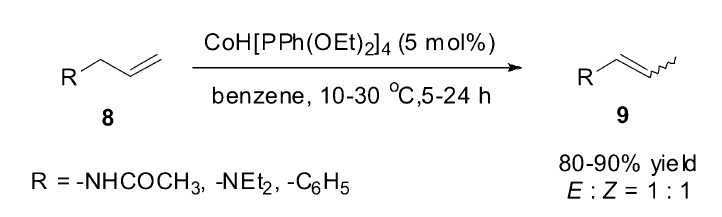 | Scheme 11 CoH[PPh(OEt)2]4catalyzed olefin isomerization[49] |
{kind=link}
1987年,Satyanarayana小组[51]研究发现,CoCl2、PPh3和NaBH4(摩尔比例1:3:1)三者混合原位产生的复合物CoHCl(PPh3)2是一个高效的烯烃异构催化剂,既可以催化直链烯烃异构,1-癸烯只迁移一个碳链生成2-癸烯,也可以高效催化烯丙基苯及黄樟脑得到完全E式产物 (Scheme 12)。
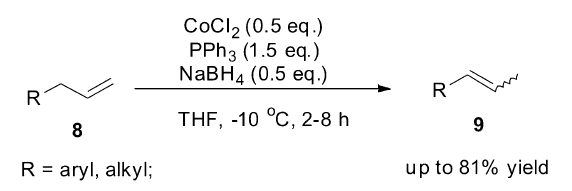 | Scheme 12 CoCl2/PPh3/NaBH4 catalyzed olefin isomerization[51] |
{kind=link}
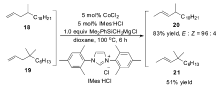
之后很长一段时间里,并没有出现新的钴催化体系,直到2009年,Oshima小组[52]在研究钴催化脱卤生成烯烃的反应中,发现延长反应时间会导致生成的烯烃在催化体系下发生异构反应,生成高度E式烯烃。 该小组经系统优化发现,以CoCl2作为催化剂,大位阻的格氏试剂作为还原剂,氮杂卡宾(NHCs)为配体,可以高选择性催化烯烃的异构反应[53],可以广泛运用于2-丁烯基和烯基硅烷的合成。 格氏试剂与配体的选择对于控制区域和立体选择性起着极其重要的作用,对于不同反应底物,也会表现出不同的催化效率。 当以4-甲基-1-十四烯(18)作为底物时,不仅需要提高格氏试剂的用量,还需要提高反应温度至100 ℃才可以得到较高的收率。 可当以烯烃19作为底物时,即使提高反应温度与格氏试剂的量,也只能以51%的收率得到异构产物(Scheme 13)。
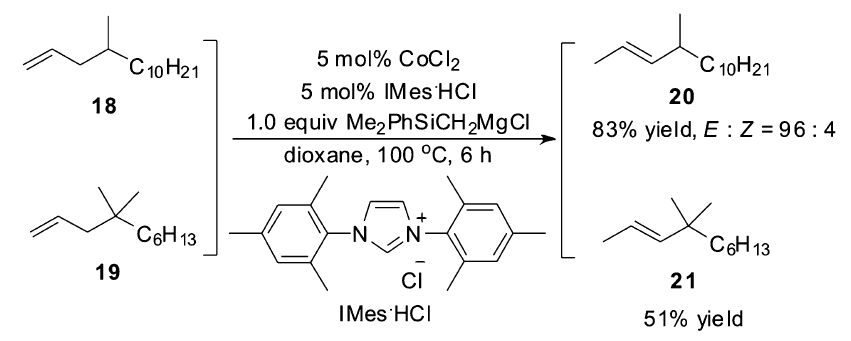 | Scheme 13 CoCl2/NHCs catalyzed olefin isomerization[53] |
{kind=link}
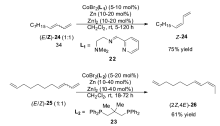
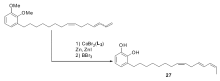
2012年之后,Hilt小组在钴催化的烯烃异构反应研究中取得了令人瞩目的成就。 2012年,该研究组发现[54],以化合物22为配体,CoBr2为催化剂,锌粉作为还原剂可以高效催化1,3-丁二烯衍生物( E/Z=1)的混合物异构生成完全 Z式的1,3-丁二烯衍生物[55],不仅适用于芳基取代的1,3-丁二烯衍生物,也适用于大量的直链1,3-丁二烯衍生物。 通常烯烃异构反应生成Z式产物是非常困难的,这对于烯烃异构反应的催化体系来说是一个很大的突破。 对该体系的后续研究发现,当以化合物23作为配体的时候,反应的活性又发生了很大的转变,1,3-丁二烯衍生物( E/Z=1)的混合物会发生双键的迁移,但是仅仅只迁移一个位置,得到2 Z,4 E-二烯化合物。 即使以化合物25作为底物时,也只会选择性反应1,3-丁二烯部分,端烯烃不会发生异构(Scheme 14)。 Hilt小组进一步将该体系成功运用于天然产物漆醇27[56]的合成(Scheme 15)。
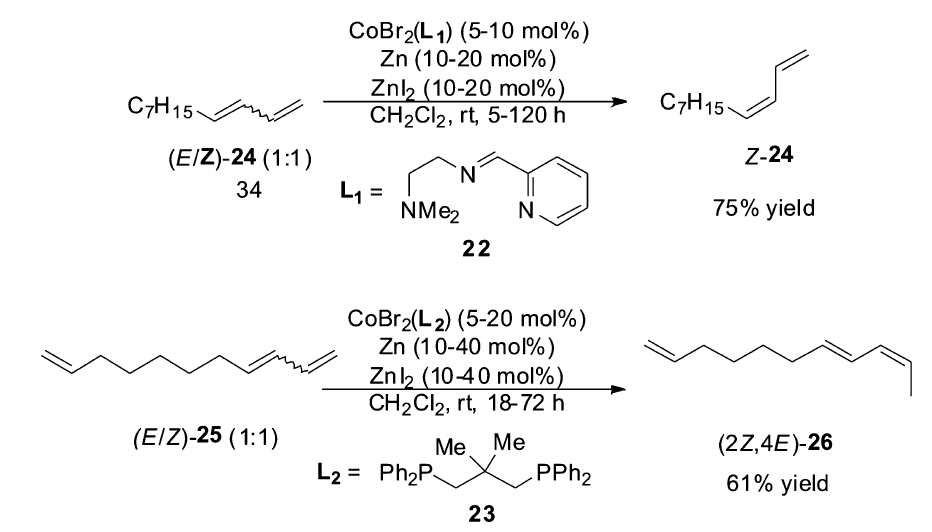 | Scheme 14 CoBr2(L)/Zn/ZnI catalyzed isomerization of 1,3-dienes[55] |
{kind=link}
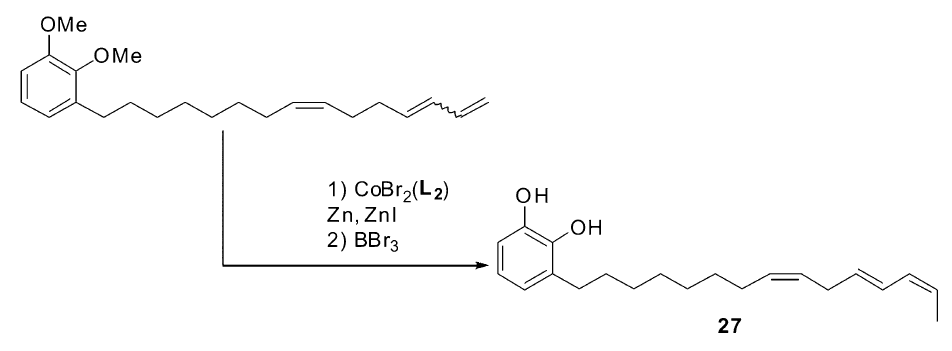 | Scheme 15 Synthesis of urushiol[56] |
{kind=link}
在实现了1,3-丁二烯衍生物的高效Z式选择性异构后,Hilt小组将钴催化体系应用到普通烯烃的 Z式选择性异构。 Z式烯烃是动力学控制的产物,Hilt小组考虑通过改变配体的位阻来控制异构产物的Z式选择性,研究发现,在反应体系中加入二苯基膦(Ph2PH),辅以合适的配体,该催化体系可以催化烯烃高区域选择性,高立体选择性异构生成 Z式产物[7](Scheme 16)。 不同的底物需要不同的配体实现最优的选择性。 当1-十六烯(28)作为底物时需要使用配体29,当5-苯基-1-戊烯(30)作为底物时需要配体31才能顺利进行。 但是对于烯丙基苯(6)作为底物时,还是无法控制立体选择性,生成完全的 E式产物。 通过氘代的二苯基膦(Ph2PD)与烯烃的反应发现,氘标记会转移至烯烃的异构产物上,而且是分子间的行为,这些证据表明,双键的迁移是通过金属钴催化下二苯基膦中的P-H对底物双键的加成消除来实现(Scheme 17)。
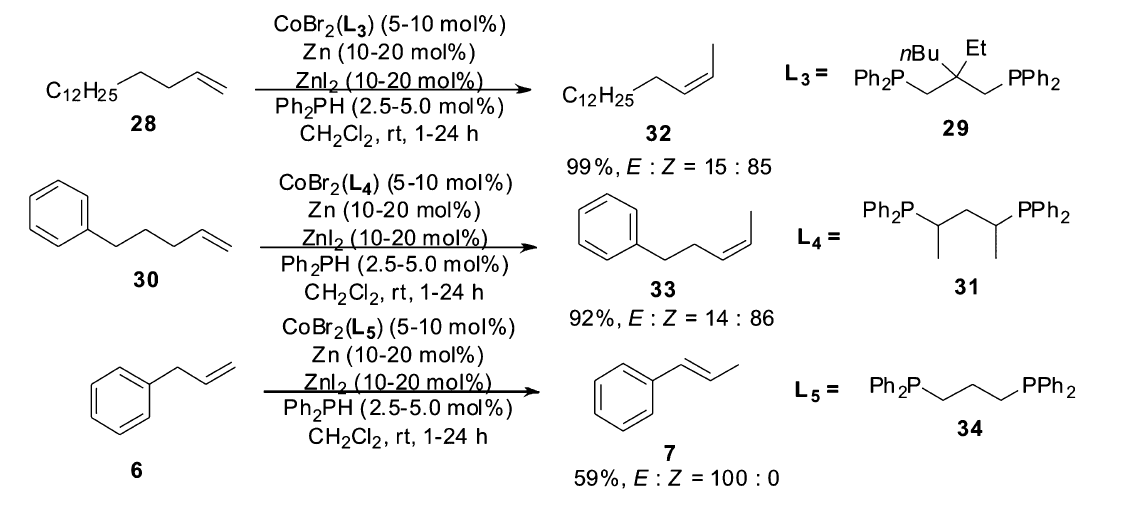 | Scheme 16 Cobalt catalyzed isomerization of terminal alkenes to ( Z)-2-alkenes[7] |
{kind=link}
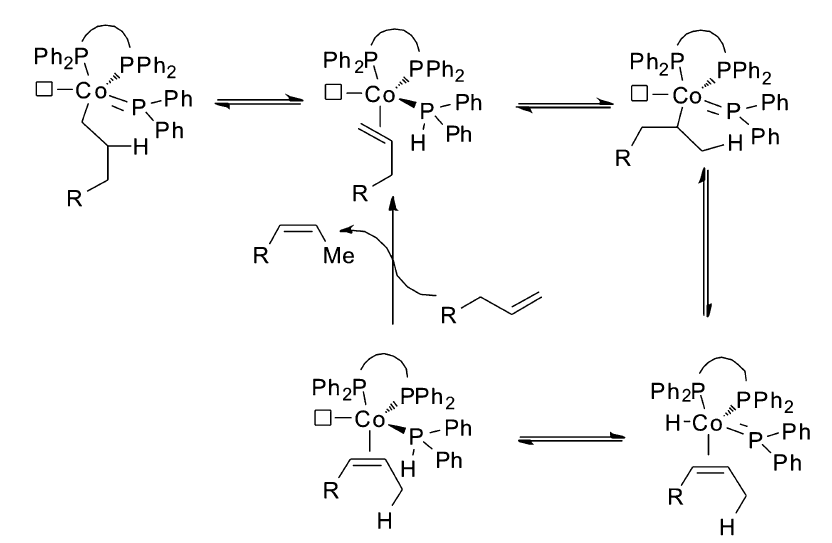 | Scheme 17 Phosphine-hydride insertion-elimination mechanism[7] |
{kind=link}
2014年,Holland小组[31]也开发出一个高效催化烯烃 Z式选择性异构的钴催化体系。 使用合成的高自旋的Co(Ⅱ)复合物35作为催化剂,可以高效催化端烯烃的 Z式选择性异构,最高的 Z/E可达到12:1。 虽然该催化体系具有高的 Z式选择性,但是还是无法控制反应的区域选择性,会出现一些双键继续沿碳链迁移的产物,而且随着时间的延长, Z式产物会一定程度上转化会热力学稳定的 E式产物。 该反应是通过1,2-加成消除机理实现双键的迁移,作者推断在 β-氢消除过程中,由于Co(Ⅱ)复合物35空间位阻效应,从而导致了产物的 Z式选择性(Scheme 18)。
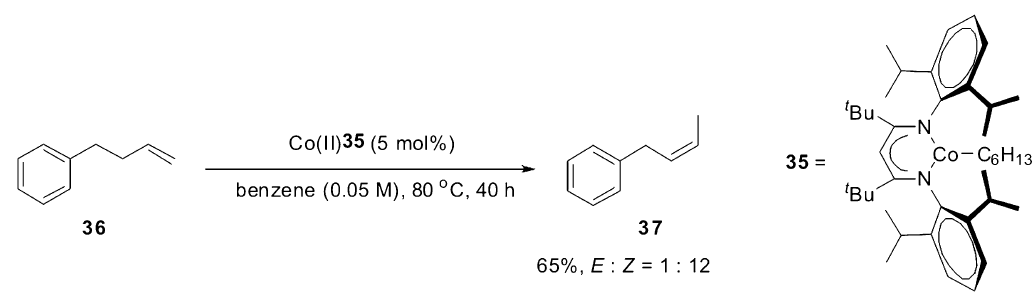 | Scheme 18 High-spin Co(Ⅱ)-complex catalyzed olefin isomerization[31] |
{kind=link}
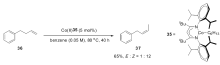
同样在2014年,Shenvi小组[57]发现了另外一个钴催化体系,以Salen钴复合物38作为催化剂,以苯基硅烷作为还原剂。 该催化体系虽然不能控制烯烃异构反应的 Z式选择性,但是可以很好地控制反应的区域选择性,双键只迁移一个位置。 该催化体系的适用范围广泛,不仅适用于普通的1-烯烃,还可以有效催化1,1-二取代烯烃的异构反应。
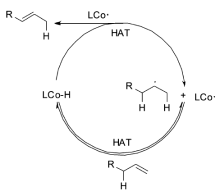
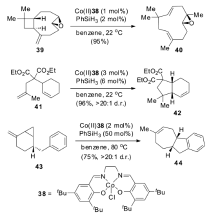
机理研究表明,该催化体系是以自由基机理来催化烯烃的异构反应,这对于过渡金属催化的烯烃异构机理来说,是一个全新的认识(Scheme 19)。 钴催化剂在还原剂存在下,首先被还原为钴氢复合物,之后与烯烃发生两次单电子转移得到烯烃异构的产物。 该催化体系被成功地用于各种大环及多环结构的合成,实现高选择性、高收率的转化(Scheme 20)。 例如,从便宜易得的石竹烯氧化物39出发一步合成40,极大简化了合成路线;从二烯化合物41出发,一步转化以96%的收率得到5,6-并环化合物42;可以顺利的将4,1,0-并环化合物43通过自由基断开环丙烷之后转化为7,5,6-三环化合物44。 这些转化极大丰富了该催化体系在合成反应中的应用。
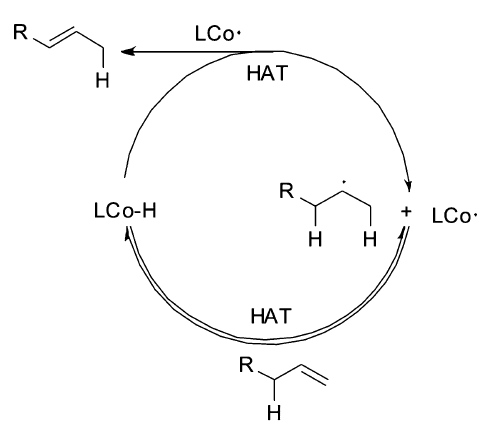 | Scheme 19 Radical mechanism for olefin isomerization[57] |
{kind=link}
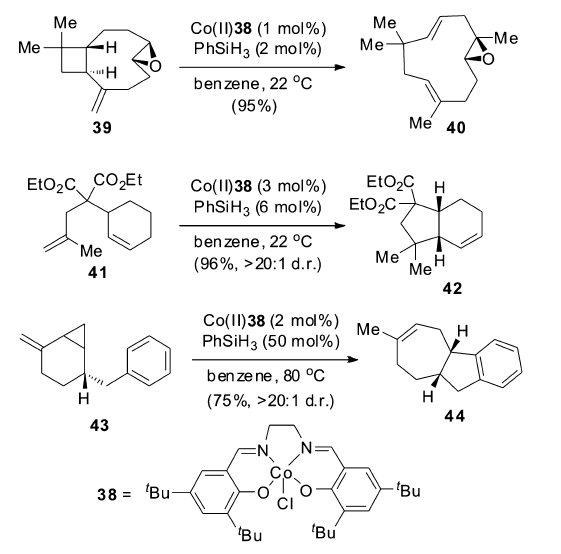 | Scheme 20 Co-salen complex catalyzed isomerization of 1,1-disubstituted alkenes[57] |
{kind=link}
由上述讨论发现,早期关于钴催化的烯烃异构反应研究比较少,集中在研究反应机理方面。 近些年,Holland小组[31]的高自旋Co(Ⅱ)复合物35以及Hilt小组[7]的CoBr2(L)/Ph2PH/Zn体系均表现出高 Z式选择性异构,这对于烯烃异构反应的选择性方面是一个重大的突破。 同时Hilt小组[7]与Shenvi小组[57]提出了不同的烯烃异构机理,增强了对过渡金属催化的烯烃异构机理的认识。
相比于金属铁和钴,镍催化的烯烃异构反应研究的较少。 1966年,Cramer小组[58]发现Ni(0)复合物Ni(POEt3)4在酸性条件下,可以在室温下快速将1-丁烯异构成为2-丁烯,而在非酸性条件下,反应无法进行,根据反应过程中1-丁烯与2-丁烯的比例变化推测的1-丁烯更容易与镍催化剂配合。 1972年,Tolman小组[59]对Ni(POEt3)4在酸性条件下催化烯烃异构反应的机理进行了更加详细的研究。 通过动力学研究发现,在酸性条件下,Ni(POEt3)4会迅速解离下一个膦配体,生成具有配位空间的Ni(Ⅰ)复合物[HNi(POEt3
1987年,Bontempelli小组[60,61]运用电化学氧化的方法处理各种Ni(0)化合物原位得到的活性镍复合物,并用于催化烯丙基苯(6)的异构反应,不同的配体对活性镍复合物的催化活性有着较大的影响。 相比于传统的化学方法,通过电化学氧化得到的活性催化物种确实具有一定的优势,但是这种催化体系缺乏广泛的适用性。
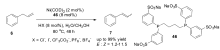
在这之后的很长一段时间里,镍催化的烯烃异构反应并没有受到太多的关注。 过去30多年来,水溶性的磷配体TPPTS(45)的出现,使得双相体系中进行金属催化反应成为可能。 与传统的有机反应相比,双相反应的催化剂后处理更加简易,更趋于一种绿色的反应过程。 1998年,Monflier课题组[62]报道了第一例水溶性的镍复合物在两相体系中催化烯烃的异构反应。以Ni(COD)2和水溶性的配体46作为催化体系,在布朗斯特酸的存在下,水相或两相体系下可高效催化烯烃的异构,布朗斯特酸对异构反应有显著的影响,当使用盐酸时,转化率可达到99%(Scheme 21)。
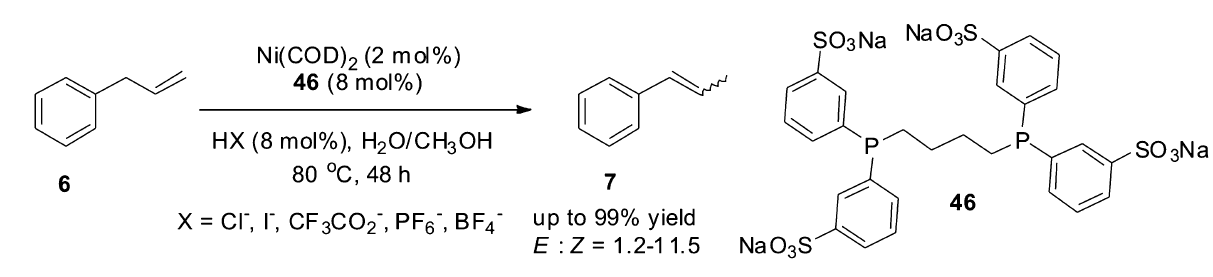 | Scheme 21 Ni(COD)2/46 catalyzed isomerization of allylbenzene[62] |
{kind=link}
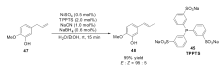
2007年,Vittori小组[63]提出以TPPTS(45)作为配体,来促进双相体系中镍催化的烯烃异构反应,研究发现Ni(0)/TPPTS体系在水相中不能稳定存在,通过筛选发现,加入一定量的NaCN,在碱性条件下可以有效稳定Ni(0)/TPPTS体系并具有很高的反应活性。 反应体系的pH值对反应影响非常大,当pH值大于11时,异构反应不会发生,在酸性条件下,催化体系不稳定,产生非活性的镍复合物。 只有当pH值处于8到11之间时,催化体系具有最高的活性。 NaCN对稳定催化体系起着至关重要的作用,但是NaCN的量过多会导致氢腈基化产物增多。 最优条件下,室温下,就可以高效催化(47)的异构反应,周转频率(turnover frequency,TOF)可达到1800 h-1,而且具有很高的立体选择性(Scheme 22)。 与之前的Monflier小组[62]的催化体系相比,该体系具有更广泛的适用范围。
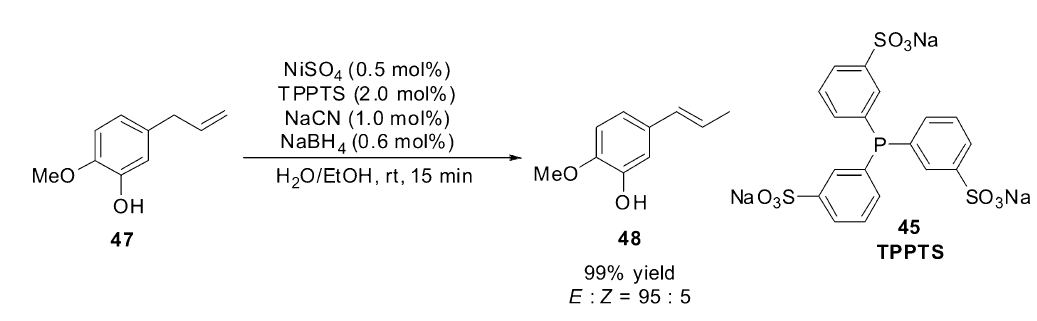 | Scheme 22 Ni(0)/TPPTS catalyzed isomerization of eugenol[63] |
{kind=link}
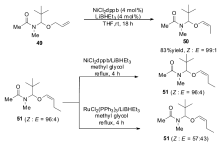
控制烯烃异构反应的立体选择性一直是研究者追求的目标。对于大多数的过渡金属催化体系,得到的都是热力学稳定的 E式烯烃,但在1998年,Frauenrath小组[64]报道了以NiCl2(dppb)作为催化剂,以LiBHEt3作为还原剂的催化体系,不仅可以高效催化烯烃的异构反应,还可以高选择性得到Z式烯烃产物。 该催化体系广泛适用于各类烯丙基醚类衍生物,当以烯烃49作为底物时, Z/E可达到99:1,尽管 E式烯烃是热力学稳定产物,但该催化体系产物的 Z/E比并不随时间延长而发生改变,而同样的底物在钌的催化体系下, Z/E比会有非常显著的下降(Scheme 23)。 该催化体系只适用于烯丙基醚类的衍生物,并不适用于其它类型烯烃的 Z式选择性异构。
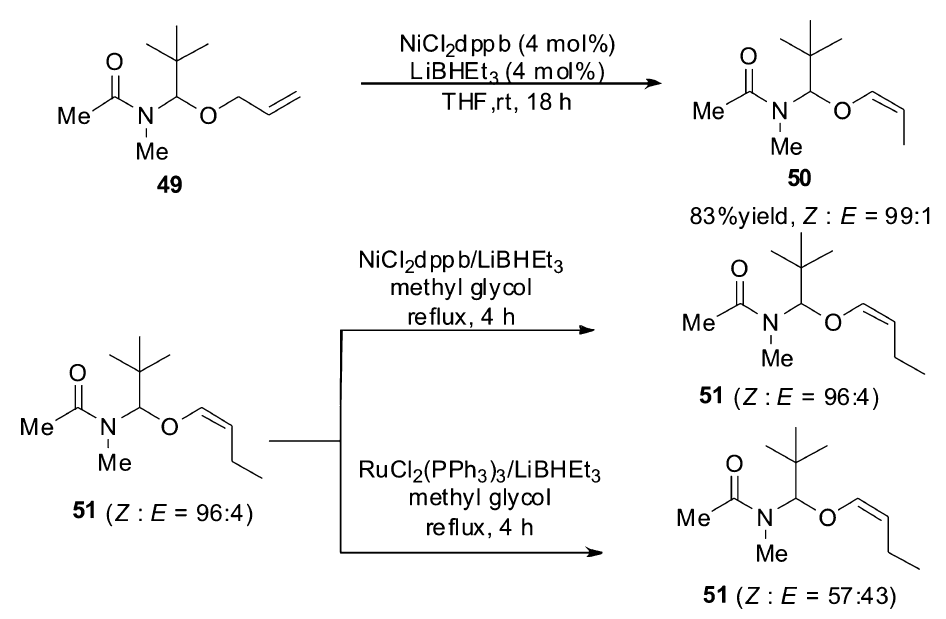 | Scheme 23 NiCl2(dppb) catalyzed isomerization of alkenes to ( Z)-alkenes[64] |
{kind=link}
2009年,RajanBabu课题组[15]在研究镍催化的乙烯对1,1-二取代烯烃的氢乙烯化反应中发现,在反应条件下烯烃原料会发生一定量的异构。 研究发现,以镍复合物[(allyl)2NiBr]2为催化剂,PPh3为配体,Na+[3,5-(CF3)2C6H3]4B-(BARF)与双烯丙基醚作为添加剂可有效将1,1-二取代烯烃52异构为产物53(Scheme 24)。 虽然可以催化1,1-二取代烯烃的异构反应,但是该催化体系与其它的催化体系相比,并没有显示出特别高的催化活性与立体选择性。
 | Scheme 24 [(allyl)2NiBr]2 catalyzed isomerization of 1,1-disubstituted alkenes[15] |
{kind=link}
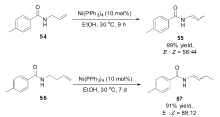
2013年,Lei小组[65]第一次报道了以镍催化的烯烃异构来合成烯胺的方法,极大地提高了烯胺的合成效率。 在研究镍催化的 α-羰基溴代烷烃的Heck偶联过程中发现, N-烯丙基-4-甲基苯甲酰胺(54)在反应体系中发生了异构反应,定量转化为烯胺化合物。 该异构反应仅需要Ni(PPh3)4作为催化剂,不需要加入任何配体和碱。 Ni(acac)2不能催化该反应,表明反应是Ni(0)催化过程,通过氘代底物在反应过程中对产物的标记研究表明反应是以分子内的1,3-氢迁移的机理进行的。 该催化体系广泛适用于多种烯丙基胺类衍生物,对于长链烯基胺类化合物56的异构反应,需要延长反应时间至一个星期,双键也可以沿着碳链迁移,最终得到烯胺类化合物(Scheme 25)。 简单的催化体系和高效的催化活性使得该催化体系具有极高的应用前景。
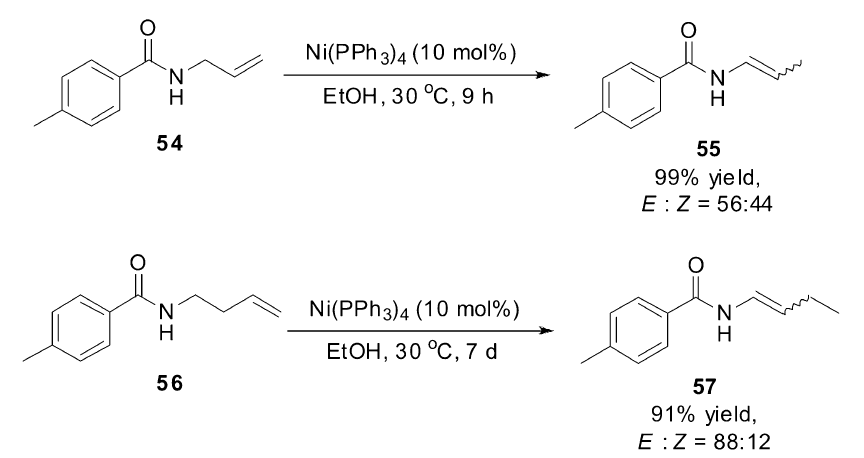 | Scheme 25 Ni(PPh3)4 catalyzed isomerization of allylamides[65] |
{kind=link}
如前文所述,Hilt小组[7]发现钴催化的烯烃异构反应中, Z式烯烃是动力学产物,降低反应的温度可以提高 Z式选择性,但是催化活性会下降。 另外对于位阻较大的烯烃,即使高温反应,钴催化剂的催化活性也不理想。 为此,Hilt小组探讨活性更高的金属镍作为催化剂诱导烯烃 Z式选择性异构的可能性[66]。 研究发现,镍复合物NiBr2(dppp)作为催化剂时,具有非常高效的催化活性,即使将反应温度降低至-60 ℃,依然可以催化烯烃58的异构,高选择性得到 Z式烯烃产物59。 该镍催化体系被成功运用于外激素前体61的合成(Scheme 26)。 该催化异构过程同样是以二苯基膦中的P-H对底物双键的加成消除来进行双键的迁移。
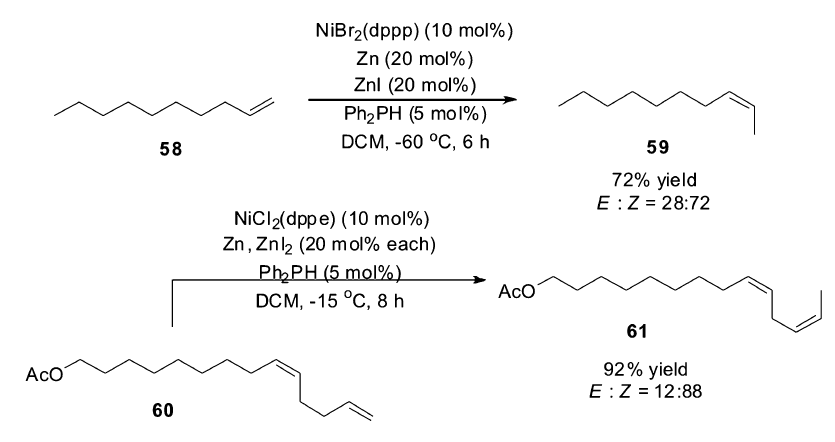 | Scheme 26 NiBr2(dppp) catalyzed isomerization of terminal alkenes to ( Z)-2-alkenes[66] |
{kind=link}
虽然经过了多年的发展,镍作为另外一个相对便宜但具有高活性的过渡金属,在烯烃异构方面的研究不是很多。 但Monflier小组[62]和Vittori小组[63]报道的两种水溶性的催化体系及Lei小组[65]的催化体系,加深了对镍催化烯烃异构反应体系的认识,将大大促进之后对镍催化烯烃异构反应体系的研究。
过渡金属催化的烯烃异构反应,由于其高效的催化活性与立体选择性,在天然产物、有机小分子、日化产品合成等领域具有广泛的应用[8,9,10,11,12]。 经过多年的发展,大量过渡金属催化体系显示出高效的催化活性。 虽然贵金属在区域选择性、立体选择性、底物的适用范围上都表现出极其优异的性能,但是廉价过渡金属催化的烯烃异构反应经过近几十年的发展,同样也可以做到同时控制好异构反应的区域和立体选择性。 不仅如此,钴和镍催化体系还实现了烯烃的Z式选择性异构,这是贵金属催化体系无法达到的,对于烯烃异构反应来说是一个很大的突破。 新的异构机理的提出改变了大家对该反应机理的固有认识,也必将引起大家对反应机理更进一步的探究,并以此设计合适的催化体系。
贵金属昂贵的价格和在医药、电子相关的合成产物中的痕量残留限制了它们更广泛的应用,这正是廉价过渡金属的优势所在。 廉价过渡金属催化的烯烃异构反应在基础研究和应用方面虽然已经取得长足的进展,展现出无与伦比的催化活性,但也面临着诸多的挑战,如何建立既具有广泛的底物适用性,又具有高效的选择性及反应活性的催化体系,如何使这些高效的催化体系适应于合成的应用,也将是廉价过渡金属催化的烯烃异构方面重要的发展方向。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|