

WU Zhiqiang, XU Zeyang, BI Shuxian, et al. Synthesis of Schiff Base Catalyzed by Coal-based Solid Acid Catalyst[J]. Chinese Journal of Applied Chemistry, 34(5): 563-571


以煤基活性炭(AC)和苯胺(ANI)为原料,通过原位-溶液聚合法制备了煤基固体酸催化剂AC@PANI-SO3H(APS),利用扫描电子显微镜(SEM)、傅里叶变换红外光谱(FTIR)、X射线光电子能谱(XPS)和热重分析(TG)等技术手段对催化剂进行了结构和性能的表征。 研究了该催化剂在微波辐射下催化合成Schiff碱化合物的活性,并对其催化工艺条件进行了优化考察。 结果表明,催化剂用量5%(以每摩尔邻苯二胺用量为基准),反应时间3~20 min,溶剂选用乙醇(EtOH),Schiff碱化合物产率可达80%~93%,说明该催化剂在微波催化合成席夫碱反应中变现出良好的催化活性,反应时间短,工艺简单操作,且催化剂能重复使用5次。 通过红外发现,催化剂重复5次后活性下降的主要原因是固体酸表面键合的磺酸基官能团消失,从而导致活性降低。
A coal-based solid acid catalyst AC@PANI-SO3H(APS) was prepared in situ by solution polymerization using coal-based activated carbon(AC) and aniline(ANI) as raw materials. The catalyst was characterized by scanning elecrton microscopy(SEM), Fourier transform infrared spectrometer(FTIR), X-ray photoelectron spectroscopy(XPS), and thermal gravimetric analyzer(TGA). Its catalytic activity on yield of Schiff bases was studied under microwave irradiation. The results show that with 5 %(based on the amount of o-phenylenediamine per mole) catalyst, the reaction in ethanol is performed for 3 to 20 min, the yield of Schiff-bases is 80% to 93%. It is proved that the catalyst exhibits good catalytic activity in the microwave-catalyzed synthesis of Schiff base. The reaction time is short, the process is simple to operate, and the catalyst can be reused 5 times. The main reason for the decrease of the activity of the catalyst after repeated 5 times is that the sulfonic acid functional groups bonded to the solid acid surface is disappeared and the activity is decreased.


席夫碱(Schiff base)是一类含有甲亚胺或亚胺基团(—R—C═N)的有机化合物,通常由伯胺与活泼羰基化合物缩合而成[1]。 席夫碱具有独特的生理和药理活性,具有抗病毒[2]、抗菌[3]、消炎[4]、抗肿瘤[5]等作用;同时,它也是有机合成的重要中间体[6]。 传统的合成方法是利用芳香醛与邻苯二胺在氧化剂或强酸催化下合成席夫碱。 该方法需较高的压力、温度或较长的时间,且副反应多、产率低[7,8]。 近年来,有文献报道利用离子液体[9]、稀有金属复合物[10]等催化制备席夫碱,该方法制备的席夫碱产率较高,但存在后处理复杂、催化剂昂贵等缺点。
碳基固体酸由于操作工艺简单,催化稳定性好和环境污染较小的优点而受到关注[11]。 宁夏太西煤具有低灰分,低S、P的特点,具备高机械强度和高化学活性等优点,是制备具有良好催化性能的材料[12,13]。 有报道[14]用MnZrO2作固体酸催化剂,酸值2.25 mmol/g,在非均相条件下合成席夫碱化合物,产品取得较好收率,但反应需要数小时。 本文以太西煤和苯胺为碳源、氮源,通过原位溶液聚合的方法制备了煤基固体酸催化剂AC@PANI-SO3H(APS,AC:活性炭,PANI:聚苯胺),用发烟硫酸气相磺化法对其进行磺化,其酸值可达1.96 mmol/g,并将其应用于微波合成席夫碱化合物的反应。 实验结果表明,催化剂APS在合成席夫碱反应中表现出很好的催化活性。 与文献报道[14,15]的固体酸相比,APS催化剂的制备及后处理简单,稳定性较好,催化活性高且能重复使用。
煤基活性炭(选用宁夏太西煤;AC,粒度0.18~0.15 mm,比表面积968.6 m2/g,总孔容0.602 cm3/g,平均孔径2.87 nm);苯甲醛(≥98.5%,北京化学试剂公司);乙腈、1,4-二氧六环(≥99.5%,上海广诺化学科技公司);苯胺(ANI)、邻苯二胺等其他试剂购自天津大茂化学试剂厂,均为分析纯。
MAS-II型微波合成/萃取仪(日本岛津公司);ASAP-2010型BET测试仪(美国Micromeritics公司);NoVaTMNano SEM-250型扫描电子显微镜(美国FEI公司);X-4型数字显示显微熔点仪(北京泰克仪器公司); FT-IR-8400型红外光谱仪(日本岛津公司);AVANCE-400型核磁共振仪(德国Bruker公司)。
采用参考文献[16]报道的方法合成碳基固体酸AC@PANI-SO3H(APS)。 将50 mL盐酸与4.0 g AC置于三口烧瓶中冰浴搅拌,待体系温度<5 ℃后,加入1.6 mL苯胺单体,搅拌20 min后,缓慢滴加3.92 g过硫酸铵水溶液,低速搅拌1 h,静置过夜,依次用盐酸、丙酮和蒸馏水洗涤至中性,80 ℃,干燥6 h,制得前驱体。 利用发烟硫酸气相磺化法在100 ℃对前驱体进行60 min的磺化。 通过酸碱中和滴定法测得酸值在1.869~1.960 mmol/g。
在25 mL的三口烧瓶中依次加入邻苯二胺1 mmol,醛1.1 mmol(或酮2.1 mmol),无水乙醇5 mL和适量催化剂(以每摩尔邻苯二胺用量为基准)催化剂,微波400 W辐射加热回流3~20 min。 反应进程用薄层色谱仪(TLC)进行跟踪( V(乙酸乙酯): V(正己烷)=1:2~1:5)。 反应结束后,混合物用10 mL CH2Cl2稀释、过滤,并用少量无水乙醇洗涤回收催化剂。 滤液用正己烷反滴重结晶或通过柱层析得到纯品。 反应式如Scheme 1所示。
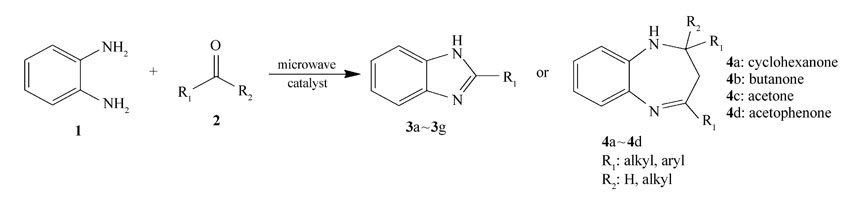 | Scheme 1 Synthetic route of Schiff bases |
{kind=link}
化合物3a:黄色固体;mp 130~133 ℃;1H NMR(400 MHz,CDCl3), δ:8.14(d, J=3.7 Hz,1H),7.89(d, J=7.9 Hz,1H),7.71(d, J=6.4 Hz,1H),7.67~7.57(m,1H),7.47(t, J=7.4 Hz,1H),7.44~7.40(m,1H),7.35(dd, J=14.7,7.3 Hz,2H),7.13(d, J=6.8 Hz,1H),5.49(s,1H);IR(KBr), σ/cm-1:3061.6(N—H),2922.2(C—H),1626.2(C═N)。
化合物3b:淡黄色固体;mp 278 ℃;1H NMR(400 MHz,CDCl3), δ:8.35~8.31(m,1H),8.27~8.22(m,1H),7.93(d, J=8.0 Hz,1H),7.87~7.81(m,1H),7.44~7.32(m,1H),7.32~7.24(m,2H),7.21(d, J=8.0 Hz,1H),5.59(s,1H),2.38(s,3H);IR(KBr), σ/cm-1:3354.9(N—H),2928.4(C—H),1638.5(C═N)。
化合物3c:黄色粉末;mp 190~191 ℃;1H NMR(400 MHz,CDCl3), δ:8.38~8.32(m,2H),8.32~8.30(m,2H),7.60~7.58(m,1H),7.56~7.53(m,1H),7.52~7.42(m,1H),7.30(d, J=8.0 Hz,1H),5.23(s,1H);IR(KBr), σ/cm-1:3289.7(N—H),2826.3(C—H),1657.6(C=N),1586.4(O-N)。
化合物3d,淡黄色粉末;mp 135~136 ℃;1H NMR(400 MHz,CDCl3), δ:8.20~8.18(m,1H),8.15~8.11(m,1H),7.59~7.58(m,2H),7.55~7.53(m,1H),7.52~7.44(m,2H),7.22(d, J=8.0 Hz,1H),4.98(s,1H);IR(KBr), σ/cm-1:3327.1(N—H),2918.4(C—H),1610.7(C=N),784.2(C—Cl)。
化合物3e,黄色固体,mp 203~205 ℃;1H NMR(400 MHz,CDCl3), δ:8.50(s,1H),8.36(dd, J=8.2,1.3 Hz,1H),8.26~8.14(m,1H),8.06(d, J=7.6 Hz,1H),7.93(d, J=7.9 Hz,1H),7.70(t, J=8.0 Hz,1H),7.56(t, J=8.0 Hz,1H),7.38(dt, J=19.8,7.1 Hz,2H),5.60(s,1H);IR(KBr), σ/cm-1:3379.3(N—H),1642.1(C=N),1545.6(-N=O)。
化合物3f,淡黄色固体;mp 253~255 ℃;1H NMR(400 MHz,CDCl3), δ:8.21~8.20(m,1H),7.95~7.93(m,1H),7.92(d, J=8.0 Hz,1H),7.84~7.80(m,1H),7.50~7.38(m,1H),7.37~7.30(m,2H),5.96(d, J=8.0 Hz,1H),5.38(s,1H);IR(KBr), σ/cm-1:3458(O—H),3263(N—H),1647(C═N)。
化合物4a:淡黄色晶体;mp 140~142 ℃;1H NMR(400 MHz,CDCl3), δ:6.63(dt, J=7.4,3.7 Hz,2H),6.59~6.53(m,2H),3.82(s,2H),1.78~1.70(m,4H),1.65~1.56(m,4H),1.45(dd, J=11.2,5.8 Hz,2H);IR(KBr), σ/cm-1:3354.9(N—H),2928.4(C—H),1493.1(C—N)。
化合物4b:淡黄色粉末,mp 134~135 ℃;1H NMR(400 MHz,CDCl3), δ:7.56(m,1H),7.12(m,1H),7.02~6.98(m,1H),6.89~6.77(m,1H),4.98(s,1H),2.98~2.87(m,4H),1.89~1.85(m,6H),1.83(m,1H) 1.56~1.44(m,4H);IR(KBr), σ/cm-1:3256.8.1(N—H),2879.6(C—H),1597.7(C═N )。
化合物4c:黄色固体;mp 121~123 ℃;1H NMR(400 MHz,CDCl3), δ:7.24(s,1H),7.08(m,1H),6.98(d, J=7.6 Hz,1H),6.77(d, J=7.9 Hz,1H),3.98(s,1H),3.22~3.20(s,2H),1.97~1.95(s,3H),1.40~1.43(s,6H);IR(KBr), σ/cm-1:3249.3(N—H),2845.3(C—H),1621.1(C═N)。
化合物4d:淡黄色固体;mp 140~142 ℃;1H NMR(400 MHz,CDCl3), δ:6.63(dt, J=7.4,3.7 Hz,2H),6.59~6.52(m,2H),3.82(s,2H),1.81~1.69(m,4H),1.65~1.55(m,4H),1.45(dd, J=11.2,5.8 Hz,2H);IR(KBr), σ/cm-1:3267.9(N—H),2928.3(C—H),1189.4(C—N)。
2.1.1 催化剂结构表征图1为催化剂磺化前后的SEM照片。 由图1 A可看出,磺化前的催化剂表面颗粒之间分散较好,但表面形貌不规整;而磺化后的催化剂(图1 B)表面形貌较为规整致密,且出现了明显的层状结构。
 | 图1 催化剂APS磺化前( A)后( B)的微观形貌Fig.1 SEM images of catalyst APS before( A) and after( B) sulphonation |
{kind=link}
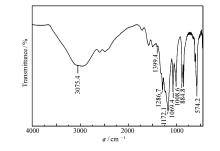
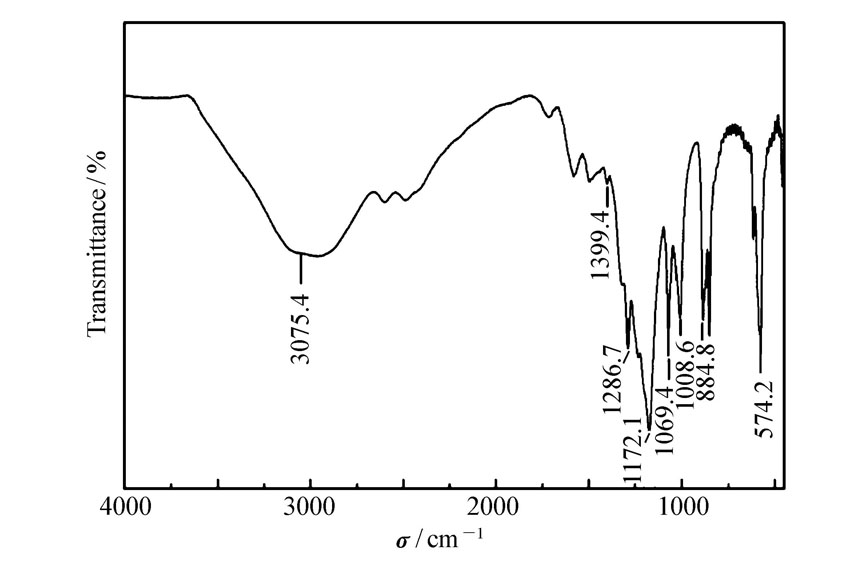 | 图2 固体酸催化剂APS的IR谱图Fig.2 IR spectra of solid acid catalyst APS |
{kind=link}
2.1.2 催化剂红外分析图2是催化剂IR表征图。 由图2可以看出,在3075.4cm-1左右出现—OH伸缩振动峰;在850~1400cm-1之间的红外特征峰证明催化剂表面含有大量硫物种的存在。 其中在884.8、1172.1 cm-1和1286.7、1399.4 cm-1处分别对应O—S—O键的对称伸缩和反对称伸缩振动峰[17];在1008.6和1069.4 cm-1处分别对应与—N2+—结构共轭和非共轭结构的苯环相连的S═O 键对称伸缩振动。 由此说明制备的催化剂与文献报道一致[16]。
2.1.3 催化剂的元素分析表1是固体酸催化剂的主要元素组成,其中S2 p特征峰的质量相对含量是6.47%,说明S元素成功的键合在前驱体表面,且由表1可得出,S/N为0.77。图3 A和3 B分别是S2 p和N1 s的XPS图谱。 从图3 A可看出,S2 p的电子结合能为168.55 eV,说明催化剂中的S元素以—SO3H基团形式存在,没有其它硫状态;从图3 B可看出,N1 s的电子结合能分别在399.55和401.5 eV,说明在催化剂中N元素呈—NH—和—N2+—价态,分别表示N元素以苯环状聚苯胺和聚苯胺盐形态的存在,因此,该催化剂的催化性能主要体现为酸催化和聚苯胺磺酸盐的协同作用。
| 表1 催化剂APS中的元素种类、结合能和原子摩尔分数 Table 1 Elemental type, binding energy and molar fraction of catalyst APS |
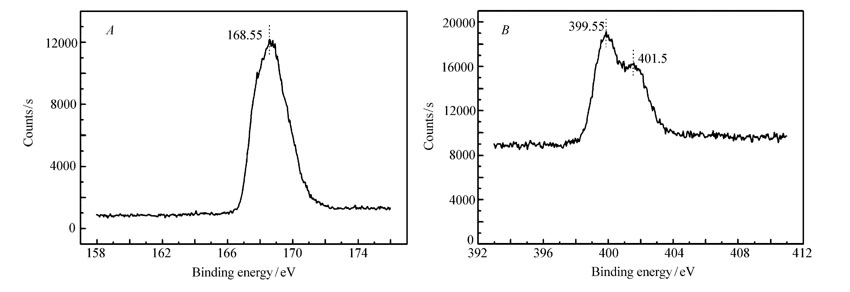 | 图3 催化剂APS中S2 p( A)和N1 s( B)的XPS能谱图Fig 3 XPS spectra of S2 p peak( A) and N1 s peak( B) in catalyst APS |
{kind=link}
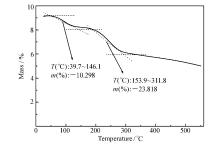
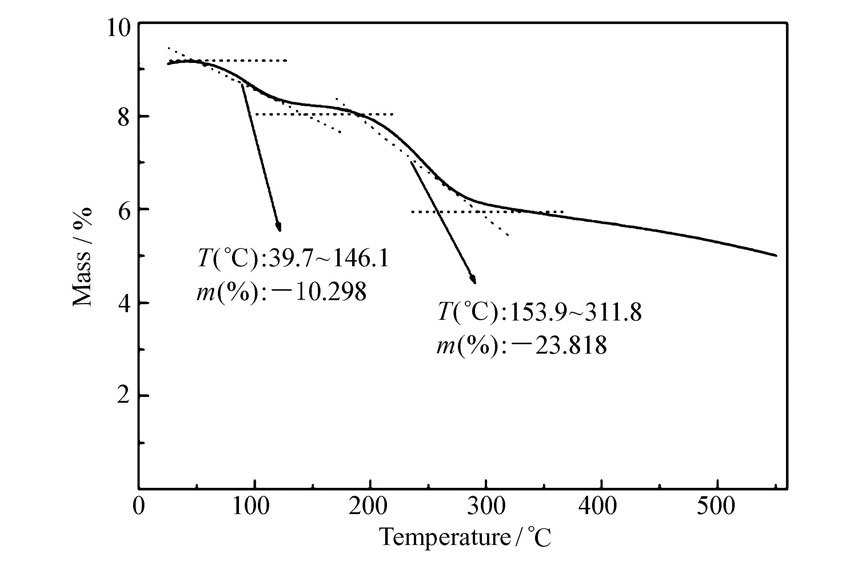 | 图4 催化剂APS的热重分析曲线Fig 4 TG curve of APS catalyst |
{kind=link}
2.1.4 催化剂的TG分析图4是催化剂APS的TG图,样品在氮气气氛受热下,分别在39.7~146.1 ℃、153.9~311.8 ℃两个区间出现失重现象。 从39.7 ℃到146.1 ℃样品质量下降约为10.3 %,其原因是固体酸催化剂中的物理吸附水;在153.9~311.8 ℃出现一个失重比较缓慢的区间,质量下降了23.8 %,其失重成分主要是掺杂态的盐酸的脱除和催化剂表面键合的—SO3H损失。
2.2.1 不同催化剂对合成席夫碱活性影响 以邻苯二胺与苯甲醛或苯乙酮反应为例,采用5种催化剂(乙酸(AcOH),三氟甲基磺酸铟(In(OTf)3),CuO-np/SiO2,三甲基氯硅烷(TMSCl)和APS)合成席夫碱化合物,考察其催化活性,实验结果见表2。 由表2可看出,不同催化剂对席夫碱的催化活性不同,其中以APS催化剂最佳,这归因于该催化剂具有较大的比表面积和微孔结构,其表面键合负载了的较多具有催化效应的磺酸基等含氧官能团。 Entry 1采用液体乙酸作催化剂在常温下合成席夫碱,反应时间短,但存在废酸量过多不易处理,并对设备造成腐蚀等缺点;Entry 2使用In(OTf)3为催化剂,反应时间较短,产品产率较高,但稀有金属的使用使得催化剂原料来源受限、价格昂贵;Entry 3和Entry 4分别使用CuO-np/SiO2和TMSCl催化剂,虽产率较高,但存在反应时间过长的缺点。 本文制备的APS煤基固体酸在微波辐射下催化合成席夫碱,不仅反应时间较短,且产品产率较高,后处理简单,说明该催化剂具有很好的催化活性。
| 表2 不同催化剂对合成席夫碱活性影响 Table 2 Effect of different catalyst activities on the synthesis of Schiff-base |
2.2.2 固体酸用量对Schiff碱产率的影响 Schiff碱的合成反应涉及到加成、重排和消去,反应物的立体结构及电子效应在合成中起到重要的作用,基于醛类比酮类活性高的考虑,实验以邻苯二胺与苯甲醛反应为例,考察催化剂用量对反应的影响,结果见表3。 可见,该合成反应在未加催化剂时,基本不发生反应;随着催化剂的加入量增大,产品收率不断提高,当催化剂用量(摩尔分数)达到5%时,产品收率最高,可达92.1%;继续增加催化剂用量,收率下降,这是因为催化剂过量会引起副反应增加,产品选择性下降。
| 表3 催化剂用量对反应的影响 Table 3 Effect of amount of catalyst on the yield |
2.2.3 反应时间对产率的影响 以邻苯二胺与苯甲醛反应为例,考察了反应时间对产率的影响,结果列于表4。 可见,随着反应时间的增加,底物与催化剂的接触时间增加,从而提高了产率。 当反应时间为10 min时,产品产率可达89.7%。 继续增加反应时间,产品产率出现下降趋势,其原因是反应时间过长,会导致副反应增加、催化剂失活或中毒等,降低了产品的选择性,最终使得产率下降。
| 表4 反应时间对产率的影响 Table 4 Effect of reaction time on the yield |
2.2.4 溶剂对产率的影响 以2.2.3节反应为例,考察溶剂对反应的影响,结果列于表5。 可见,随着溶剂的极性增大,Schiff碱收率逐渐提高,其原因是[21]溶剂极性的增大降低了该反应物、中间体和产物的溶剂化能,提高了碳正离子中间体和产物的生成,同时促进化合物的稳定,从而提高产品产率。 采用乙腈作溶剂时,产品收率达92.6%,这是因为乙腈作为一种极性较强的含氮有机溶剂,它对原料和产物均有很好的溶解性,可以促进反应的平衡,从而提高产品收率,但由于乙腈有毒,对环境造成污染。 故实验采用相对环保的无水乙醇作为反应溶剂,其产率可达到87.4%。
| 表5 溶剂条件对产率的影响 Table 5 Effect of solvents on the yields |
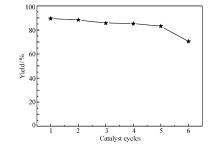
2.2.5 催化剂重复使用性 以邻苯二胺与苯甲醛反应为例,考察催化剂的重复使用性。用1 mmol邻苯二胺与1.1 mmol苯甲醛投料比反应,催化剂用量5 %,反应时间10 min,无水乙醇为溶剂,待反应结束后,采用二氯甲烷溶解混合物,过滤催化剂,并用无水乙醇洗涤,80 ℃干燥6 h后直接用于下一次反应,催化剂重复性见图5。 由图5可以看出,该催化剂在重复5次后,催化活性明显下降。
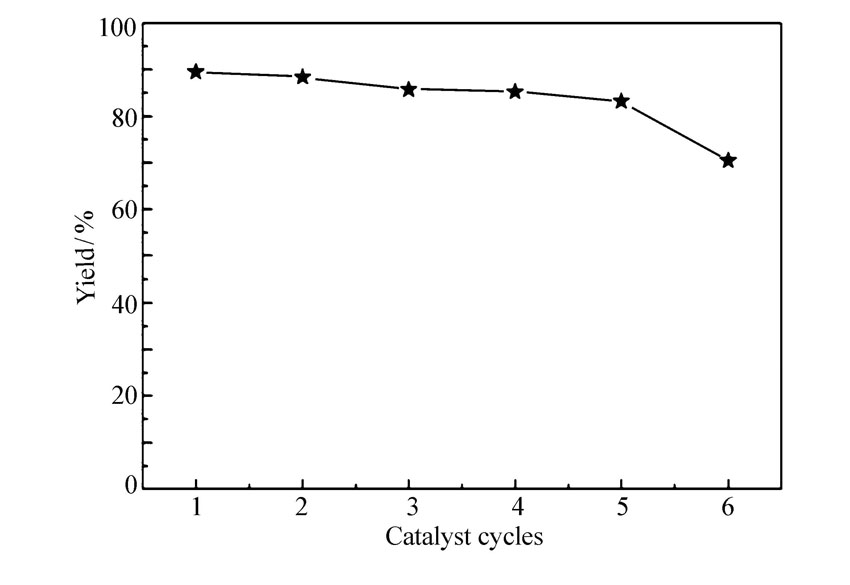 | 图5 催化剂APS重复使用性Fig.5 Reusabilities of the catalyst APS |
{kind=link}
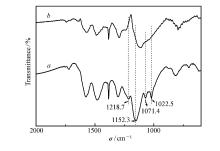
图6为催化剂在反应前与反应6次后的IR对比图。 可见,催化剂在重复使用6次后,其表面的O—S—O特征峰和S═N特征峰消失,表明催化剂表面的—SO3H消失,导致催化剂酸值降低,从而使得催化活性下降。
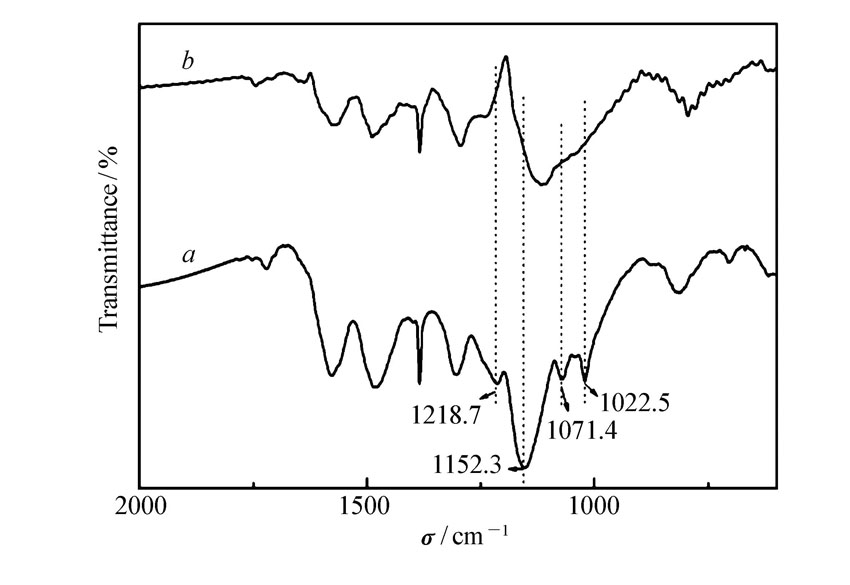 | 图6 催化剂APS反应1次后( a)和6次后( b)的IR谱图Fig.6 IR spectra of catalyst APS after one( a) and six( b) time reactions |
{kind=link}
2.2.6 煤基固体酸催化不同底物合成Schiff碱 在上述的优化工艺下,考察APS催化邻苯二胺与醛(酮)在微波辐射下合成席夫碱化合物,结果列于表6。 可见,邻苯二胺与醛或酮均能发生反应,这是因为羰基为亲核加成提供了活性位点,但由于受到空间位阻效应和溶剂极性大小的影响,不同取代基对应不同的合成产率。 其中化合物3a~3f的产率高于化合物4a~4d,原因可能是醛基相连的取代基体积明显小于酮基相连的取代基体积。 当氮原子进攻羰基上的碳原子后,碳原子的电子轨道由 sp2杂化变为 sp3杂化,化学键由原来的120°缩小为109.5°,取代基体积变小,空间位阻变小,有利于化学反应的进行。 因此,醛比酮的空间位阻相对要小,醛类更易于发生反应。
| 表6 煤基固体酸APS微波辐射下催化合成Schiff碱 Table 6 Preparation of Schiff-base catalyzed by carbon based solid acid APS under microwave |
2.2.7 催化合成反应机理 在酸性条件下,Schiff碱的合成机理如(Scheme 2);第一步是羰基的质子化,在—SO3H作用下,胺与醛(酮)上的羰基发生亲核加成,去质子使得氮上的电子发生转移,接着脱去一分子H2O,从而得到一个亚胺离子中间体。 第二步是醛(酮)在酸性条件下生成烯醇中间体,亚胺离子作为亲核试剂,进攻具有活泼氢的化合物的烯醇型结构,质子失去,从而得到产物。
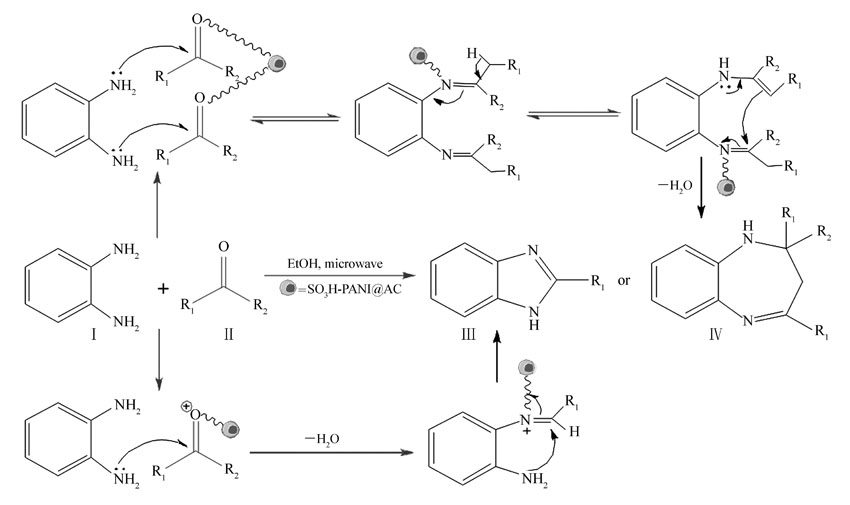 | Scheme 2 Reaction mechanism for catalytical synthesis of Schiff bases |
{kind=link}
以太西煤为碳源,通过原位-溶液聚合法,制备了较高酸密度的煤基固体酸催化剂,将该催化剂应用于微波合成席夫碱的研究,考察其工艺参数。 分析比较了APS固体酸、液体酸、有机酸和负载型质子酸催化剂对催化席夫碱反应的活性研究。 结果表明,在微波辐射条件下,APS催化剂表现出较好的催化活性,反应时间大大缩短,产率明显得到提高。 此外,对席夫碱反应的工艺参数进行了优化,当催化剂用量(摩尔分数)为5%;反应时间3~20 min;无水乙醇作溶剂,席夫碱产率可达80~93%,催化剂重复5次后催化活性无明显下降,说明该催化剂具有良好的催化活性和稳定性。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|