
LI Zhao, FENG Jia, GAO Yuanyuan, et al. Synthesis of Key Impurities as Quality Control Standards of Oxfendazole[J]. Chinese Journal of Applied Chemistry, 33(9): 1061-1066


共同通讯联系人:朱维良,研究员; Tel:021-50805020; E-mail:wlzhu@simm.ac.cn; 研究方向:药物设计与合成
苯并咪唑氨基甲酸酯类化合物(Benzimidazolecarbamate,BZC)是一类广谱抗寄生虫抗菌剂,其广泛使用的典型代表药物为奥芬达唑。 但该药物中的杂质化合物不易获得,导致生产质控不足。 本文发现了奥芬达唑中4种主要杂质成分,包括芬苯达唑、5-苯磺酰基-1 H-2-甲氧甲酰氨基苯并咪唑、5-苯亚磺酰基-1 H-2-氨基苯并咪唑和2-二(5-苯亚磺酰基-1 H-2-苯并咪唑基)-碳酰二胺的简单高效化学合成方法。 特别是通过两种不同的关环策略实现了关键吡唑环的有效合成,以及控制氧化条件得到硫醚的亚砜和砜的氧化产物。 该方法不仅可为奥芬达唑的质控提供标准品,而且为基于奥芬达唑类似物的新型杀虫、抗菌及抗肿瘤等药物的研发提供了一条简单高效的合成线路。
Co-corresponding author:ZHU Weiliang, professor; Tel:021-50805020; E-mail:wlzhu@simm.ac.cn; Research interests:drug design and synthesis
Oxfendazole, a benzimidazolecarbamate(BZC) compound, is a broad-spectrum antihelmintic and antimicrobial agent. The insufficient sources of its impurities affected the quality control of the product. Herein, a simple and efficient method for synthesis of four impurities, fenbendazole, methyl (5-(phenylsulfonyl)-1 H-benzo[d]imidazol-2-yl)carbamate, 5-(phenylsulfinyl)-1 H-benzo[d]imidazol-2-amine and 1,3-bis(5-(phenylsulfinyl)-1 H-benzo[d]imidazol-2-yl)urea, is revealed. Especially, the key imidazole ring was constructed efficiently via two different strategies. Furthermore, oxidative products of sulfur ether, sulphone and sulfoxide were obtained by controlling oxidative conditions. This method not only increases the source of the standard substances for quality control of oxfendazole, but also provides an efficient synthetic strategy to prepare the novel antihelmintic, antimicrobial and antitumor agents based on oxfendazol.
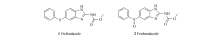
苯并咪唑氨基甲酸酯类化合物(Benzimidazolecarbamate,BZC)是一类广谱抗蠕虫制剂[1,2],对线虫、绦虫、吸虫均具有很好的杀灭和抑制效果[3],还有报道发现这类制剂同时具有抗肿瘤[4]和抗真菌[5]的作用。 其广泛的药理作用,可能与其抑制微管蛋白(Tubulin)[6]以及细胞色素P450[7]有关。 BZC的代表性化合物为奥芬达唑(Oxfendazole,1)和芬苯达唑(Fenbendazole,2)(图1),临床上主要用于牛、羊、猪、马等家畜肠道寄生虫的治疗。 奥芬达唑与芬苯达唑的抗寄生虫谱基本一致,但是前者具有更强的活性,从上市以来一直得到广泛的应用。
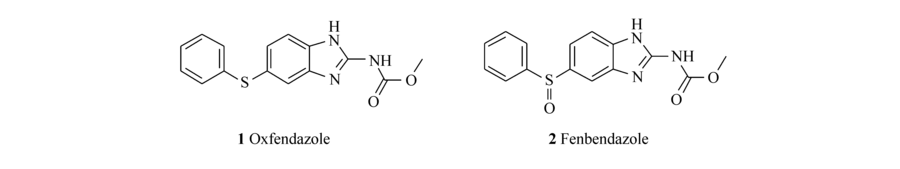 | 图1 苯并咪唑氨基甲酸酯类代表性化合物Fig.1 Representative compounds of benzimidazole carbamate |
{kind=link}
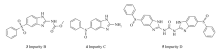
《中国兽药典》2010年版规定奥芬达唑有关物质的检测采用薄层色谱法,而《英国兽药典》早在2003年版就采用了高效液相色谱法。 其中,《英国兽药典》收录的奥芬达唑的杂质主要有:杂质A(芬苯达唑,2)(图 1)、杂质B(5-苯磺酰基-1 H-2-甲氧甲酰氨基苯并咪唑,3)、杂质C(5-苯亚磺酰基-1 H-2-氨基苯并咪唑,4)、杂质D(2-二(5-苯亚磺酰基-1 H-2-苯并咪唑基)-碳酰二胺,5)(图 2)。 造成我国《兽药典》没有采用奥芬达唑有关物质高效液相色谱法检测的主要原因之一是除杂质A外,其它杂质均不易购买,且价格昂贵[8]。
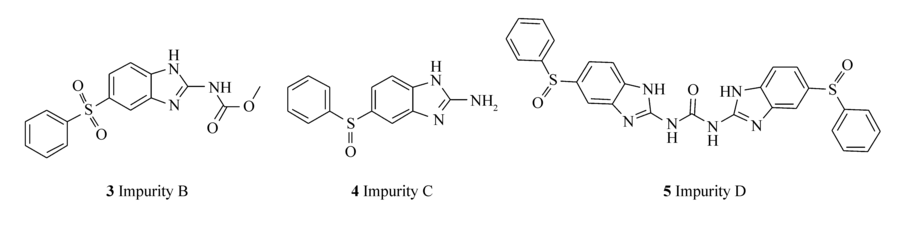 | 图2 奥芬达唑中的杂质成分B、C、DFig.2 Impurities B, C and D in oxfendazole |
{kind=link}
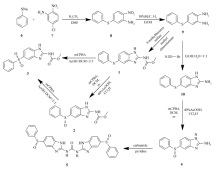
并且,除了杂质A和B外,杂质C和D目前没有相关合成方法报道。 因此,针对以上问题,我们研究开发了奥芬达唑杂质A、B、C、D的合成方法(Scheme 1)。 该方法可用于大规模生产,成本低廉,收率较高,可以解决奥芬达唑质控中杂质对照品不易获得的难题。
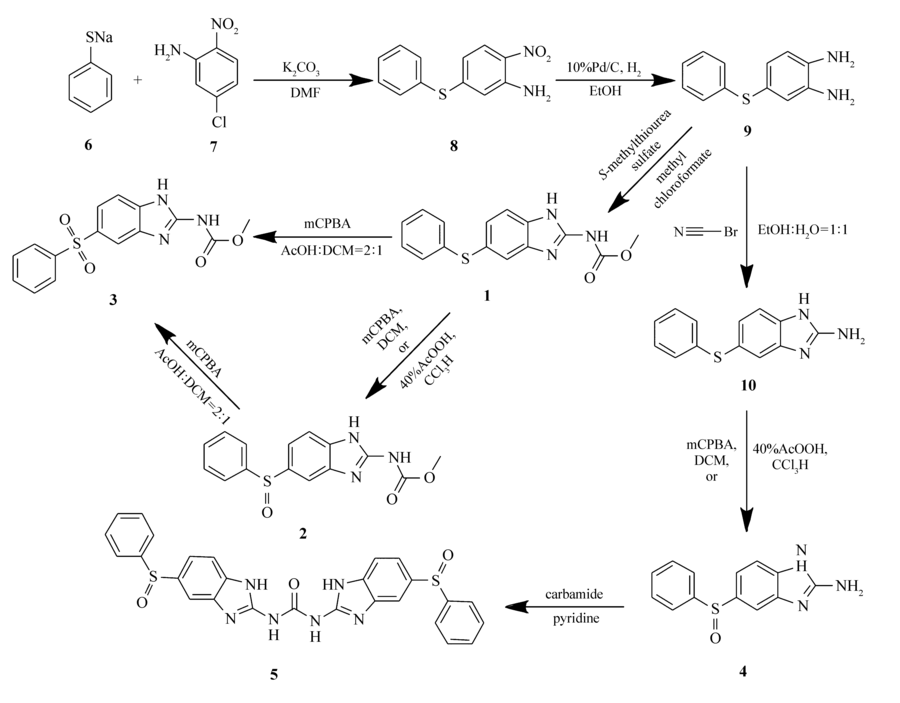 | Scheme 1 The synthetic route of impurities |
{kind=link}
Agilent 1260型高效液相色谱仪(美国Agilent公司);Bruker Avance-400或500型核磁共振仪(美国Bruker公司);LCQ-DECA型质谱仪(德国Finnigan公司,ESI-MS);Waters Q-TOF Ultima型质谱仪(美国Waters公司,HR-ESI-MS);实验所用试剂均为市售化学纯或者分析纯。
1.2.1 2-硝基-5-苯硫基苯胺(化合物8)的制备 将苯硫酚钠6(1.32 g,12.0 mmol)与5-氯-2-硝基苯胺7(1.73 g,10.0 mmol)溶于30 mL DMF中,反应体系用N2气置换,再加入K2CO3(1.38 g,10.0 mmol)于上述反应液中,90 ℃加热反应36 h。 反应完全后,将反应液加250 mL水稀释,有黄色固体析出,过滤,弃滤液,滤饼用水洗并且60 ℃真空干燥12 h,得棕黄色固体:2.20 g,收率:89%。1H NMR(400 MHz,Chloroform-d), δ:8.00(d, J=9.05 Hz,1H),7.62~7.51(m,2H),7.50~7.40(m,3H),6.44(d, J=9.08 Hz,1H),6.38(s,1H),6.05(s,2H);ESI-MS:245.1[M+1]+[9]。
1.2.2 4-苯硫基-1,2-二氨基苯(化合物9)的制备 将化合物8(1.00 g,4.1 mmol)与10%(质量分数)Pd/C(0.10 g)溶于20 mL无水乙醇中,反应体系用H2气置换,50 ℃加热反应24 h。 反应完全后,将反应液用硅藻土过滤,滤饼用乙醇洗,合并滤液减压浓缩,得棕色浓稠油状液体:822.7 mg,收率:94%。1H NMR(400 MHz,Chloroform-d), δ:7.29~7.09(m,5H),6.95~6.85(m,2H),6.71(d, J=7.88 Hz,1H),3.54(s,2H),3.40(s,2H);ESI-MS:217.3[M+1]+[9]。
1.2.3 5-苯硫基-1 H-2-甲氧甲酰胺基苯并咪唑(化合物1)的制备 将 S-甲基异硫脲硫酸盐(65.3 mg,0.35 mmol)溶于1 mL水中,冰浴,待温度降低到0 ℃后再加入氯甲酸甲酯(51.6 μL,0.67 mmol),并缓慢滴加25%(质量分数)NaOH(0.11 g,2.77 mmol)于上述反应液中,反应40 min,并逐渐升至室温。 将25%(质量分数)冰醋酸水溶液(1.00 mL,16.66 mmol)加入到上述反应液中,再迅速加入溶于乙醇(1.5 mL)的化合物9(60 mg,0.28 mmol),反应液升温至95 ℃加热反应24 h。 反应完全后,反应液加水稀释,二氯甲烷萃取3次,有机相合并,水洗,饱和食盐水洗,无水硫酸钠干燥,减压浓缩,残余物硅胶柱层析分离( V(二氯甲烷): V(甲醇)=40:1),得淡黄色固体:25.0 mg,收率31%。1H NMR(400 MHz,DMSO-d6), δ:11.98(s,1H),11.42(s,1H),7.52(s,1H),7.45(d, J=7.27 Hz,1H),7.29(t, J=7.66 Hz,2H),7.22~ 7.09(m,4H),3.76(s,3H);ESI-MS:300.2[M+1]+[9]。
1.2.4 5-苯亚磺酰基-1 H-2-甲氧甲酰胺基苯并咪唑(化合物2)的制备 将化合物10(20 mg,0.067 mmo)溶于醋酸(2 mL)与二氯甲烷(1 mL)的混合溶液中,冰浴。 待温度降低至0 ℃后,再加入间氯过氧苯甲酸mCPBA(11.5 mg,0.067 mmol),反应3 h,并逐渐升至室温。 反应完全后,反应液加饱和碳酸氢钠水溶液适量,二氯甲烷萃取3次,有机相合并,水洗,饱和食盐水洗,无水硫酸钠干燥,减压浓缩,残余物硅胶柱层析分离( V(二氯甲烷): V(甲醇)=30:1),得淡黄色固体:5.3 mg,收率25%。1H NMR(400 MHz,DMSO-d6), δ:11.87(s,2H),7.75(s,1H),7.67(d, J=7.52 Hz,2H),7.51(dq, J=5.31,6.83,12.82 Hz,4H),7.39(d, J=8.27 Hz,1H),3.77(s,3H);13C NMR(125 MHz,DMSO-d6), δ:149.30,147.15,131.13,130.10,129.78,124.48,53.09;ESI-MS:316.1[M+1]+。
1.2.5 5-苯磺酰基-1 H-2-甲氧甲酰氨基苯并咪唑(化合物3)的制备 将化合物2(3.15 g,10.0 mmol)溶于200 mL醋酸和100 mL二氯甲烷的混合溶液中。 再加入mCPBA(6.92 g,40.0 mmol),室温下反应过夜。 反应液加饱和碳酸钠溶液淬灭,有大量不溶固体析出,过滤,弃滤液,滤饼用二氯甲烷洗,水洗,真空干燥,得白色固体:1.36 g,收率40%。1H NMR(400 MHz,DMSO-d6), δ:7.96(s,1H),7.90(d, J=7.55 Hz,2H),7.66~7.45(m,5H),3.75(s,3H);13C NMR(100 MHz,DMSO-d6), δ:155.33,142.79,132.92,131.59,126.78,119.67,114.12,113.29,52.27;ESI-MS:332.1[M+1]+。
1.2.6 5-苯硫基-1 H-2-氨基苯并咪唑(化合物10)的制备 将化合物9(253.8 mg,1.17 mmol)与溴化氰(131 mg,1.24 mmol)溶于5 mL乙醇和5 mL水的混合溶液中,70 ℃加热反应1 h。 反应液降低到室温后,滴加1 mol/L氢氧化钠水溶液调pH=10,再加入过量饱和硫代硫酸钠水溶液,减压蒸去乙醇,残液用二氯甲烷萃取3次,有机相合并,水洗,饱和食盐水洗,无水硫酸钠干燥,减压浓缩,残余物硅胶柱层析分离( V(二氯甲烷): V(甲醇)=40:1),得棕黄色固体粉末:231.9 mg,收率96%。1H NMR(400 MHz,DMSO-d6), δ:10.86(s,1H),7.28~7.19(m,3H),7.18~7.09(m,2H),7.09~6.99(m,3H),6.39(s,2H);13C NMR(125 MHz,DMSO-d6), δ:156.86,140.04,129.46,127.25,126.30,125.76,120.50;ESI-MS:242.3[M+1]+。
1.2.7 5-苯亚磺酰基-1 H-2-氨基苯并咪唑(化合物4)的制备 将化合物10(1.00 g,4.3mmol)与溴化氰(0.98 g,9.2 mmol)溶于30 mL乙醇和30 mL水的混合溶剂中。 70 ℃加热反应1 h。 反应液降低至室温后,滴加1 mol/L氢氧化钠水溶液调pH=10,再加入过量饱和硫代硫酸钠溶液,1 mol/L盐酸水溶液调pH=7,减压浓缩,残余物硅胶柱层析分离( V(二氯甲烷): V(甲醇)=20:1)。 得棕黄色固体粉末:0.89 g,产率84%。1H NMR(400 MHz,DMSO-d6), δ:10.96(s,1H),7.63(d, J=7.27 Hz,2H),7.49(dt, J=7.02,13.83 Hz,3H),7.37(s,1H),7.19(t, J=6.23 Hz,2H),6.50(s,2H);13C NMR(125 MHz,DMSO-d6), δ:157.11,146.97,130.38,129.18,123.90;ESI-MS:258.2[M+1]+。
1.2.8 2-二(5-苯亚磺酰基-1 H-2-苯并咪唑基)-碳酰二胺(化合物5)的合成 将化合物4(2.17 g,8.4 mmol)溶于10 mL吡啶中,再加入脲素(0.33 g,5.5 mmol),135 ℃加热回流反应20 h。 停止加热,将吡啶减压蒸干,再加甲醇超声,过滤,滤饼用甲醇洗,弃滤液,得白色固体:1.25 g,收率57%。1H NMR(400 MHz,DMSO-d6), δ:12.09(s,4H),7.54(s,2H),7.48(d, J=7.88 Hz,2H),7.35~7.27(m,4H),7.27~7.12(m,8H);13C NMR(125 MHz,DMSO-d6), δ:147.07,146.57,146.43,131.22,129.83,124.49;ESI-MS:538.9[M-1]-。
反应路线最长为5步反应,采取不同的咪唑关环和硫醚氧化策略可分别得到不同的目标产物(Scheme 1)。 首先,我们以苯硫酚钠6和5-氯-2-硝基苯胺7为起始原料,以K2CO3作碱,在DMF中加热90 ℃反应得到苯硫醚中间体8。 然后再将中间体8用10%Pd/C催化,利用氢气还原硝基,得到中间体9。 然后将 S-甲基异硫脲硫酸盐与氯甲酸甲酯溶于水并滴加25%NaOH水溶液,在冰水浴中反应1 h,逐渐升至室温,再在上述反应液中加入过量的25%醋酸水溶液,然后迅速加入中间体9的乙醇溶液,95 ℃下加热反应24 h,得到奥芬达唑中间体1。 由中间体1制备终产物2可以用1倍化学计量的mCPBA或者40%(质量分数)的过氧乙酸氧化得到。 而终产物3可以用5倍化学计量的间氯过氧苯甲酸氧化得到。 我们也可以通过将终产物2用过量的间氯过氧苯甲酸继续氧化得到终产物3。 将中间体9与溴化氰在乙醇和水的混合溶液中,60 ℃加热反应可以得到2-氨基苯并咪唑中间体10。将中间体10进一步利用1倍化学计量的间氯过氧苯甲酸或者40%(质量分数)过氧乙酸反应,可以得到终产物4。 再将4与脲素溶于吡啶,在135 ℃下回流反应20 h得到终产物5。
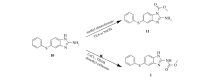
我们还尝试利用中间体10来制备中间体1,多次优化了文献[10-11]报道的利用碳酸二甲酯或者氯甲酸甲酯制备氨基甲酸酯的方法,却均未得到中间体1。 其中,以碳酸二甲酯作为试剂和反应溶剂在尝试CaCl2和四丁基溴化铵不同化学计量配比下均未得到产物1(Scheme 2)。 而利用氯甲酸甲酯在不同的碱作为缚酸剂(比如:三乙胺、氢氧化钠)下反应仅仅得到咪唑环上N甲酰化的产物11(Scheme 2)。
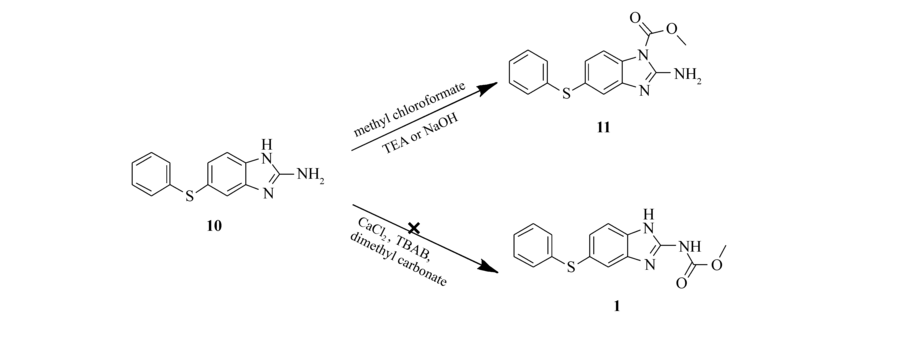 | Scheme 2 Optimization of synthetic conditions of carbamate |
{kind=link}
在以化合物4为原料制备杂质D(化合物5)的过程中,我们筛选了一系列的羰基供体、碱以及溶剂(表1)。 对于合成化合物5这样的对称型碳酰二胺,比较理想的试剂应该是三光气,但是在筛选了二氯甲烷和四氢呋喃这两个溶剂后,发现利用三光气并不能反应获得目标产物(表1,反应1、2)。 有文献[12]报道, N, N-羰基二咪唑(CDI)可以用于2-氨基吡啶对称性酰胺的合成,我们发现,在以DMF作溶剂三乙胺作碱的情况下,反应液80 ℃反应24 h后出现沉淀。 经过滤并用水和乙醇等溶剂洗涤后得白色粉末,但该白色粉末几乎不溶于任何溶剂。 最后通过ESI以及用氘代DMSO作为溶剂测定NMR1H谱,结果表明,这白色的粉末即是目标产物(杂质D)。 但该合成路线的收率很低(~1%,表1,反应4)。 因此,十分有必要对该合成路线开展优化研究。我们首先尝试了1,8-二氮杂双环[5.4.0]十一碳-7-烯(DBU)和N, N-二甲基-4-吡啶胺(DMAP)作碱,但反应都没有获得目标产物(表1,反应5、6)。 考虑到脲素的分解温度为135 ℃,我们研究了脲素在吡啶作溶剂时,直接加热到135 ℃的反应情况,结果是以57%的高收率获得目标产物(表1,反应9)。 这个方法不仅产率高,而且纯化十分方便,仅需减压浓缩将反应液中的吡啶除去,然后残余物加甲醇混悬过滤,滤饼用适量甲醇清洗即可得到白色粉末状固体的化合物5。
| 表1 杂质5合成条件的筛选 Table 1 Optimization of synthetic conditions of impurity D |
综上所述,我们报道了奥芬达唑杂质成分A、B、C、D的新合成方法。 利用两种不同的咪唑关环和硫醚氧化策略实现了杂质A、B及C(即化合物2、3、4)的合成,以及利用脲素在135 ℃下分解用于杂质D(化合物5)的合成。 该方法反应条件温和,所用试剂易得,产物纯化简便,反应收率高,可用于大量制备,不仅可为奥芬达唑的质量控制提供标准品,而且为基于奥芬达唑类似物的新型杀虫、抗菌及抗肿瘤等药物的研发提供了一条简单高效的合成线路。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|