
CHEN Meijun, LI Jinhai, ZHANG Zhijia, et al. Synthesis and Anti-tumor Evaluation of Nitrile-Containing Gambogic Acid Derivatives[J]. Chinese Journal of Applied Chemistry, 33(8): 905-912


以藤黄酸为原料,经过酯化反应或酰胺化反应,在C-30位的羧基上引入不同的烷氰基或芳香氰基,设计合成了7个藤黄酸氰基衍生物,其中6个为新化合物,其结构经MS和1H NMR确证。 采用四氮唑蓝(MTT)法测试了合成化合物对肝癌细胞(HepG2)、结肠腺癌细胞(RKO)和卵巢腺癌细胞(OVCAR-3)的体外抗肿瘤活性,结果表明,所合成的化合物均具有一定的抗肿瘤活性,其中化合物4和6的抗肿瘤活性明显优于阳性对照物藤黄酸。
Seven nitrile-containing gambogic acid derivatives were synthesized from gambogic acid(GA) through introducing different alkylnitriles or arylnitriles on the C-30 carboxyl of gambogic acid. Six of them are new compounds(1~6). Their structures were identified by1H NMR and MS analysis. The evaluation of antitumor bioactivities in vitro on the hepatoma cells(HepG2), colon adenocarcinoma cells(RKO) and ovarian adenocarcinoma cells(OVCAR-3) were done by the methylthiazolydiphenyl-tetrazolium bromide(MTT) method. The results show that most of the synthesized compounds have strong antitumor activities. Among them, the antitumor activities of compounds 4 and 6 are more potent than that of gambogic acid.
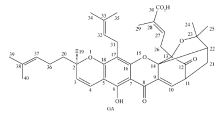
藤黄酸(GA)为双子叶植物药藤黄科植物藤黄中提取的主要活性成分,是一种桥环氧杂蒽酮类天然药物(图1)[1,2,3]。 藤黄酸已经被国家食品药品监督管理总局(CFDA)批准作为多靶点抗肿瘤药物[4,5,6,7]进行Ⅱ期临床试验[8,9],其能够选择性地抑制结肠癌、肝癌等肿瘤细胞的增殖[10,11,12],具有诱导细胞凋亡和抑制通道蛋白表达的抗肿瘤作用[13,14],且对体内正常细胞的毒性作用极小。 作为一种新型的天然多靶点抗肿瘤药物,其在临床实践中具有良好的应用前景[8,9]。 但是,由于GA具有复杂特异的4-氧杂-三环并[4.3.1.03,7]癸-2-酮笼状桥环的化学结构,对热碱不稳定,易发生环裂解,具有水溶性小、稳定性差的缺点,在临床上的应用受到限制。 因此,有必要对GA分子结构进行适当的改造,使其具有更好的药理学活性和水溶性。
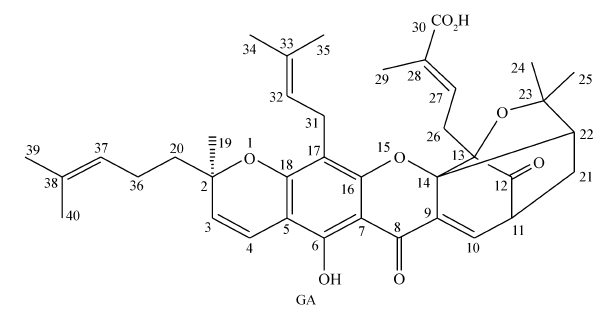 | 图1 藤黄酸的化学结构式Fig.1 Structure of gambogic acid |
{kind=link}
由于氰基具有独特的理化性质,它在药物的结构修饰与改造中扮演着重要的角色。 氰基具有较强的极性,是一个良好的氢键受体,因此,在药物分子中引入氰基可以有效地改善药物的溶解度,提高生物利用度;氰基具有较小的体积(体积仅为甲基的12.5%),因此能通过较小的空隙深入到靶标蛋白深处与活性部位的氨基酸残基形成氢键相互作用;同时,氰基还是羰基、卤素等多种基团的生物电子等排体,还可以作为羟基、羧基的替代基团;由于氰基是强吸电子基团,在芳环上的氰基取代基可以抑制氧化代谢,提高化合物在体内代谢的稳定性[15,16,17]。 鉴于氰基的这些特殊性质,在小分子药物中引入氰基已成为药物结构修饰改造和结构优化的一个重要策略。 目前,临床上使用的许多药物中含有氰基基团,如痛风性关节炎治疗药物非布索坦[18]、前列腺癌治疗药物比卡鲁胺[19]、艾滋病治疗药物依曲韦林和利匹韦林等[15]。
近年来,人们对藤黄酸衍生物进行了较广泛的研究[20,21,22,23],在这些研究的基础上,本文以增加藤黄酸的抗肿瘤活性、克服其水溶性和稳定性差等缺点为目的,在C-30位的羧基上通过酯键或酰胺键引入烷氰基或芳香氰基基团,设计合成了7个藤黄酸的氰基衍生物,化合物结构经1HNMR、MS确证。
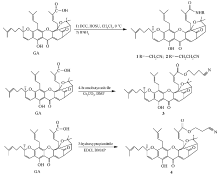
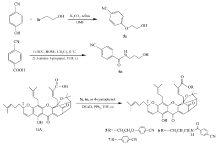
化合物1~4是具有不同碳链长度的藤黄酸氰烷基酯或酰胺。 GA分别与氨基乙腈和3-氨基丙腈在偶联剂二环己基碳二亚胺(DCC)/ N-羟基琥珀酰亚胺(HOSU)作用下得到藤黄酸氰烷基酰胺1和2;GA与4-溴丁腈在碳酸铯的催化作用下进行亲核取代反应得到藤黄酸氰基醇酯3;GA与3-羟基丙腈在偶联剂1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDCI)/4-二甲氨基吡啶(DMAP)存在下反应得到藤黄酸氰基醇酯4(见Scheme 1)。 化合物5~7是含芳香氰基的藤黄酸衍生物,它们由GA分别与化合物5a、6a或4-氰基苯酚通过光延反应得到。 化合物5a由4-氰基苯酚与2-溴乙醇在无水碳酸钾存在下反应生成;化合物6a由4-氰基苯甲酸与3-氨基-1-丙醇偶联得到(见Scheme 2)。
目标产物的生物活性测试采用四氮唑蓝(MTT)还原法,以GA为阳性对照,测试它们对肝癌细胞HepG2、结肠腺癌细胞RKO、卵巢腺癌细胞OVCAR-3的体外抗肿瘤活性。
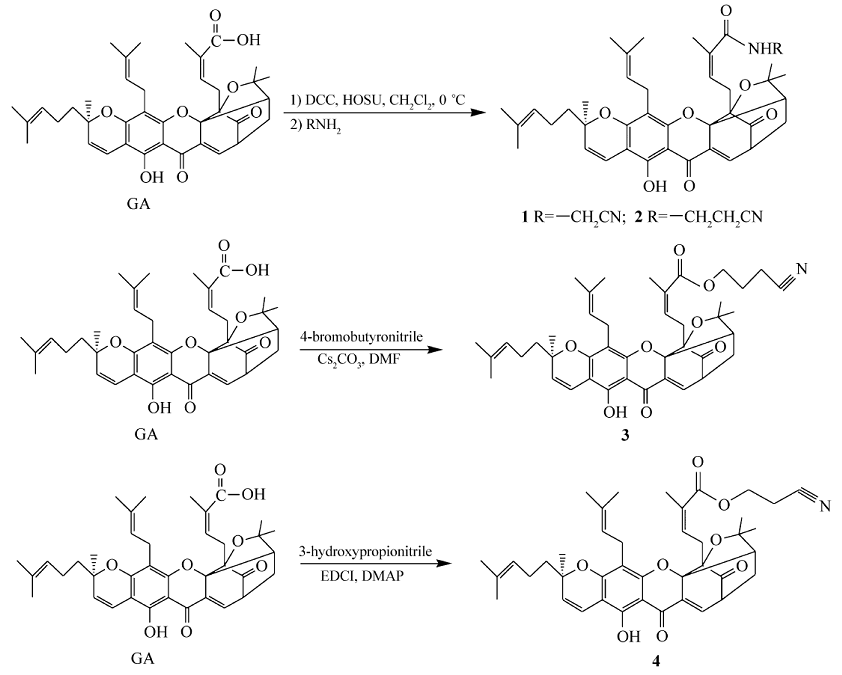 | Scheme 1 The synthetic routes of target products 1~4 |
{kind=link}
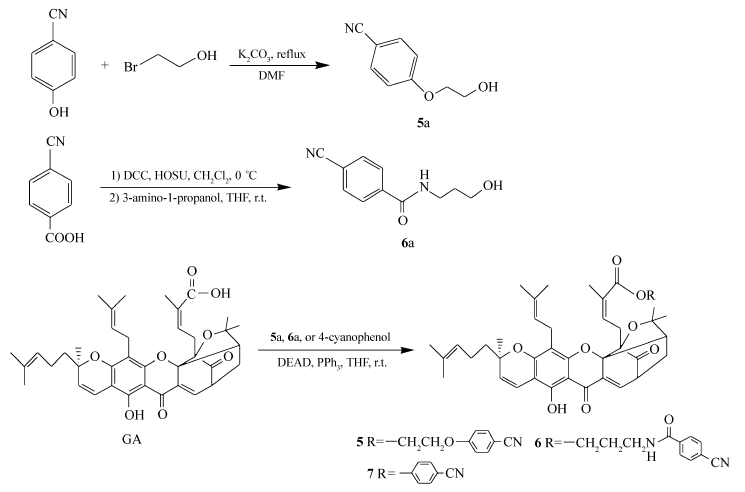 | Scheme 2 The synthetic routes of target products 5~7 |
{kind=link}
Bruker 400MHz型核磁共振仪(德国Bruker公司);Varian Mercury-Plus 300型核磁共振仪(美国Varian公司);LC MS-2010A型液相色谱质谱联用仪(日本岛津公司);Agilent 6330型质谱仪(美国Agilent公司);Tecan M1000型酶标仪(瑞士Tecan公司);Thermo Scientific 8000系列CO2细胞培养箱(美国Thermo Scientific公司)。
藤黄原料(广州清平中药材市场,实验室提纯,纯度大于95%);TLC薄层层析硅胶GF254(青岛海洋化工厂);柱层析硅胶(青岛海洋化工厂);1640培养基(GIBCO);胎牛血清(杭州四季青公司);四甲基唑氮蓝(MTT,Sigma公司);其余实验用试剂均为市售分析纯(国药集团化学试剂有限公司)。
1.2.1 藤黄酸的提取 提取工艺都是参考S.A.Ahmad的传统方法[24]:首先,藤黄粉末溶于丙酮混合加热提取3次(温度控制在60 ℃)得粗提物;然后,粗提物和吡啶成盐制备成粗品藤黄酸吡啶盐,粗品藤黄酸吡啶盐在甲醇中3次重结晶得精制藤黄酸吡啶盐;最后,将精制的藤黄酸吡啶盐溶解于乙醚并用两倍于精制藤黄酸吡啶盐摩尔量的盐酸游离得到藤黄酸单体。
1.2.2 N-腈甲基藤黄酰胺(化合物1)的合成 将藤黄酸(200 mg,0.32 mmol)和HOSU(74 mg,0.64 mmol)溶于15 mL干燥的二氯甲烷中,于0 ℃冰浴中充分搅拌混溶,30 min后,用10 mL的CH2Cl2溶解DCC(130 mg,0.64 mmol)成DCC溶液,然后于0 ℃下逐滴滴加DCC溶液,自然升至室温,室温搅拌1 h,过滤,浓缩,再加入氨基乙腈盐酸盐(120 mg,1.27 mmol)溶于10 mL的THF中,加入少量碳酸铯至反应液中继续搅拌,反应9 h。 乙酸乙酯萃取,饱和食盐水水洗3次,分出乙酸乙酯层,无水硫酸钠干燥,过滤,旋干浓缩,得到橘黄色胶状产物。 柱层析[ V(石油醚): V(乙酸乙酯)=4:1]纯化,得淡黄色粘稠物(化合物1)114 mg,收率为53%。1H NMR(CDCl3,300 MHz), δ:12.80(s,1H,6-OH),11.9(s,1H,CONH),7.60(d, J=7.6 Hz,,1H,10-H),6.69(d, J=14.3 Hz, 1H,4-H),5.49(t, J=9.03 Hz,1H,27-H),5.16(m,1H,3-H),5.09(m,2H,32-H,37-H),3.54(m,1H,11-H),3.32(m,2H,31-H),3.07(m,2H,26-H),2.85(s,2H,CONHCH2CN),2.64(m,1H,22-H),2.12(m,2H,21-H),1.98(s,2H,36-H),1.79(s,3H,25-H),1.74(s,3H,29-H),1.68(s,5H,20-H,34-H),1.61(s,6H,35-H,39-H),1.43(m,3H,40-H),1.39(s,3H,24-H),1.28(s,3H,19-H);ESI-MS m/z:667[M+H]+(计算值 m/z:666.3)。
1.2.3 N-氰乙基藤黄酰胺(化合物2)的合成 将藤黄酸(200 mg,0.32 mmol)及HOSU(74 mg,0.64 mmol)溶于15 mL干燥的二氯甲烷中,于0 ℃的冰浴中充分搅拌混溶,30 min后,用10 mL的CH2Cl2溶解DCC(130 mg,0.64 mmol)成DCC溶液,然后于0 ℃下逐滴滴加DCC溶液,自然升至室温,室温下搅拌1 h,过滤,浓缩,再加入3-氨基丙腈(0.052 mL,0.64 mmol)溶于15 mL的THF中,反应8 h。 乙酸乙酯萃取,饱和食盐水水洗3次,分出乙酸乙酯层,无水硫酸钠干燥,过滤,旋干浓缩,得到橘黄色胶状产物。 柱层析[ V(石油醚): V(乙酸乙酯)=5:1]纯化,得淡黄色粘稠物(化合物2)133 mg,收率为61%。1H NMR(CDCl3,300 MHz), δ:11.69(s,1H,6-OH),7.56(d, J=6.9 Hz,1H,10-H),6.90(s,1H,CONH),6.68(d, J=11.0 Hz,1H,4-H),5.49(d, J=10.3 Hz, 1H,3-H),5.08(m,2H,32-H,37-H),4.96(s,1H,27-H),3.71(m,2H,CONHCH2CH2CN),3.56(m,1H,11-H),3.49(m,2H,CONHCH2CH2CN),3.22(m,2H,31-H),2.85~2.75(m,2H,26-H),2.58(m,1H,22-H),2.33(m,2H,21-H),2.11(s,1H,36a-H),2.06(s,1H,36b-H),1.74(s,3H,25-H),1.72(s,3H,29-H),1.66(s,5H,20-H,34-H),1.65~1.56(m,6H,39-H,35-H),1.40(s,3H,40-H),1.26(s,6H,24-H,19-H);ESI-MS m/z: 681[M+H]+(计算值 m/z:680.3)。
1.2.4 藤黄酸丁腈酯(化合物3)的合成 将藤黄酸(200 mg,0.32 mmol)与无水碳酸铯(310 mg,0.96 mmol)溶于10 mL的二甲基甲酰胺(DMF)中,室温下搅拌,30 min后,用10 mL的DMF溶解4-溴丁腈(280 mg,1.91 mmol)成4-溴丁腈溶液,然后在搅拌下滴加4-溴丁腈溶液,反应24 h。 乙酸乙酯萃取,饱和食盐水水洗3次,分出乙酸乙酯层,无水硫酸钠干燥,旋干浓缩,得到橘黄色产物。 柱层析[ V(石油醚): V(乙酸乙酯)=4:1]纯化,得淡黄色粘稠物(化合物3)127 mg,收率为57%。1H NMR(CDCl3,300 MHz), δ:12.93(s,1H,6-OH),7.59(d, J=6.9 Hz,1H,10-H),6.58(s,1H,4-H),6.00(t, J=7.5 Hz,1H,27-H),5.24(m,1H,3-H),5.16~5.09(m,2H,32-H,37-H),4.02~3.96(m,2H,COOCH2CH2CH2CN),3.51(m,1H,11-H),3.36(m,4H,COOCH2CH2CH2CN),3.10~3.04(m,2H,31-H),2.96~2.90(m,2H,26-H),2.53(d, J=9.5 Hz,1H,22-H),2.36(m,2H,21-H),2.10(m,2H,36-H),1.83(s,3H,25-H),1.79(s,3H,29-H),1.73(s,5H,20-H,34-H),1.70(s,3H,39-H),1.61(s,6H,35-H,40-H),1.30(s,3H,24-H),1.27(s,3H,19-H); ESI-MS m/z:696[M+H]+(计算值 m/z:695.3)。
1.2.5 藤黄酸丙腈酯(化合物4)的合成 将藤黄酸(200 mg,0.32 mmol)溶于4 mL的3-羟基丙腈中,加入DMAP(80 mg,0.64 mmol)与EDCI(121 mg,0.64 mmol)后室温下搅拌反应16 h,直至原料药反应完全后停止反应。 乙酸乙酯萃取,饱和食盐水水洗3次,分出乙酸乙酯层,无水硫酸钠干燥,旋蒸浓缩,得到橘黄色产物。 柱层析[ V(石油醚): V(乙酸乙酯)=4:1]洗脱,得淡黄色胶状物(化合物4)100 mg,收率为46%。1H NMR( CDCl3,300 MHz), δ:12.79(s,1H,6-OH),7.56(d, J=6.8 Hz,1H,10-H),6.59(d, J=10.2 Hz, 1H,4-H),6.08(t, J=7.6 Hz,1H,27-H),5.40(d, J=10.1 Hz, 1H,3-H),5.04(m,2H,32-H,37-H),4.33(d, J=4.36 Hz,2H,COOCH2CH2CN),3.77(s,2H,COOCH2CH2CN),3.48(m,1H,11-H),3.31(m,2H,31-H),2.93(m,2H,26-H),2.51(d, J=9.5 Hz,1H,22-H),2.29(m,2H,21-H),2.03(m,2H,36-H),1.74(s,3H,25-H),1.72(s,3H,29-H),1.64(s,2H,20-H),1.62(s,3H,34-H),1.58(s,3H,39-H),1.54(s,3H,35-H),1.38(s,3H,40-H),1.25(s,6H,24-H,19-H);ESI-MS m/z:682[M+H]+(计算值 m/z:681.3)。
1.2.6 藤黄酸-2-(4-氰基苯氧基)乙酯(化合物5)的合成 1)将4-氰基苯酚(477 mg,4 mmol)溶解于10 mL的无水DMF中,然后加入2-溴乙醇(1000 mg,8 mmol)和无水碳酸钾(1106 mg,8 mmol),加热回流16~20 h,直到反应结束。 用乙酸乙酯稀释混合液,过滤,过滤后用饱和食盐水洗,无水硫酸钠干燥,蒸出溶剂,柱层析[ V(石油醚): V(乙酸乙酯)=3:1]洗脱,得白色固体粉末5a,390 mg,产率60%。1H NMR(CDCl3,400 MHz), δ:2.00(s,1H,—OH),4.00(s,2H,1-H),4.13(s,2H,2-H),6.97(d, J=7.90 Hz,2H,Ar—H),7.58(d, J=7.90 Hz,2H,Ar—H);ESI-MS m/z:186[M+Na]+(计算值 m/z:163.1)。
2)将藤黄酸(250 mg,0.4 mmol)溶于10 mL无水THF中,加入产物5a(131 mg,0.8 mmol)和三苯基磷(210 mg,0.8 mmol),室温搅拌15 min,然后逐滴加入用3 mL无水THF稀释的偶氮二甲酸二乙酯(139 mg,0.8 mmol),室温反应24 h,直到反应结束,浓缩,柱层析[ V(石油醚): V(乙酸乙酯)=2:1]洗脱,得橙黄色固体(化合物5),200 mg,产率65%。1H NMR(CDCl3,400 MHz), δ:1.28(s,6H,19-H,24-H),1.34(s,1H,21b-H),1.39(s,3H,40-H),1.57(s,6H,35-H,39-H),1.64~1.68(m,8H,20-H,34-H,29-H),1.73(s,3H,25-H),1.95~2.06(m,2H,36-H),2.28(d, J=13.56 Hz,1H,21a-H),2.50(d, J=8.90 Hz, 1H,22-H),2.91~3.05(m,2H,26-H),3.05~3.21(m,2H,31-H),3.27~3.41(m,1H,11-H),4.06(s,2H,1'-H),4.15~4.21(m,2H,2'-H),5.03(s,2H,32-H,37-H),5.40(t, J=10.12 Hz,1H,3-H),6.02~6.11(m,1H,27-H),6.62(d, J=10.32 Hz,1H,4-H),6.89(d, J=7.08 Hz, 2H,Ar—H),7.31~7.34(m,1H,10-H),7.58(m,2H,Ar-H),12.85(s,1H,6-OH);ESI-MS m/z:796[M+Na]+(计算值 m/z:773.4)。
1.2.7 藤黄酸-3-(4-氰基苯甲酰氨基)丙酯(化合物6)的合成 1)将4-氰基苯甲酸(590 mg,4 mmol)溶解于15 mL的二氯甲烷中,加入 N-羟基琥珀酰亚胺(905 mg,6 mmol),在0 ℃中混溶,然后加入用3 mL二氯甲烷溶解的二环己基碳二亚胺(1238 mg,6 mmol),混合自然反应至室温,室温反应1 h,过滤,浓缩,加入3-氨基丙醇(451 mg,6 mmol),然后用10 mL的THF溶解,在室温下反应15 h,直到反应结束,过滤,乙酸乙酯萃取,分出乙酸乙酯层,饱和食盐水水洗,无水硫酸钠干燥,旋干浓缩,柱层析[ V(石油醚): V(乙酸乙酯)=1:1]洗脱,得白色固体粉末6a,280 mg,产率35%。1H NMR(DMSO,400 MHz), δ:1.65~1.71(m,2H,2-H),1.99(2,1H,—OH),3.30~3.33(m,2H,3-H),3.44~3.48(d, J=11.4 Hz,6.3Hz,2H,1-H),7.97(m,4H,Ar—H),8.68~8.70(t, J=5.5 Hz,1H,—NH);ESI-MS m/z:227[M+Na]+(计算值 m/z:204.1)。
2)将藤黄酸(189 mg,0.3 mmol)溶于10 mL无水THF中,加入产物6a(110 mg,0.5 mmol)和三苯基磷(158 mg,0.6 mmol),室温搅拌15 min,然后逐滴加入用3 mL无水THF稀释的偶氮二甲酸二乙酯(105 mg,0.6 mmol),室温反应24 h,直到反应结束,浓缩,柱层析[ V(石油醚): V(乙酸乙酯)=1:1]洗脱,得橙黄色固体(化合物6)180 mg,产率72%。1H NMR(CDCl3,400 MHz), δ:1.25~1.30(m,4H,19-H,24-H),1.35(s,1H,21b-H),1.41(s,3H,40-H),1.58(s,6H,35-H,39-H),1.64(s,5H,20-H,34-H),1.70(s,6H,29-H,25-H),1.76~1.82(m,2H,2'-H),1.93~2.11(m,2H,36-H),2.32(d, J=12.75 Hz,1H,21a-H),2.53(d, J=8.50 Hz,1H,22-H),3.17(s,2H,26-H),3.25~3.39(m,3H,31-H,11-H),3.45~3.52(m,2H,1'-H),3.97~4.12(m,2H,3'-H),5.03(s,2H,32-H,37-H),5.43(t, J=10.32 Hz,1H,3-H),6.08~6.16(m,1H,27-H),6.59(s,1H,4-H),7.51~7.55(m,1H,10-H),7.76(m,2H,Ar—H),7.92(m,2H,Ar—H),12.90(s,1H,6-OH);ESI-MS m/z:815[M+H]+(计算值 m/z:814.4)。
1.2.8 藤黄酸-4'-氰基苯酯(化合物7)的合成 将藤黄酸(189 mg,0.3 mmol)溶于5 mL无水THF中,加入4-氰基苯酚(72 mg,0.6 mmol)和三苯基磷(158 mg,0.6 mmol),室温搅拌15 min,然后逐滴加入用3 mL无水THF稀释的偶氮二甲酸二乙酯(105 mg,0.6 mmol),室温反应24 h,直到反应结束,浓缩,柱层析[ V(石油醚): V(乙酸乙酯)=2:1]洗脱,得棕黄色固体(化合物7)110 mg,产率50%。1H NMR(CDCl3,400 MHz), δ:1.27(s,6H,19-H,24-H),1.38(s,3H,40-H),1.59(s,6H,35-H,39-H),1.63(s,3H,34-H),1.66(s,2H,20-H),1.73(s,3H,29-H),1.88(s,3H,25-H),2.01~2.04(m,2H,36-H),2.36(m,2H,21-H) 2.55(d, J=8.90 Hz, 1H,22-H),3.02(m,2H,26-H),3.29(m,2H,31-H),3.50(m,1H,11-H),5.04(s,2H,32-H,37-H),5.43(d, J=10.12 Hz,1H,3-H),6.30~6.39(m,1H,27-H),6.62(d, J=10.32 Hz,1H,4-H),7.03(d, J=7.08 Hz,2H,Ar—H),7.52~7.54(m,1H,10-H),7.61(m,2H,Ar—H),12.79(s,1H,6-OH);ESI-MS m/z:730[M+H]+(计算值 m/z:729.3)。
1.2.9 体外抗肿瘤活性测定 采用MTT法,以藤黄酸为阳性对照物,测定目标化合物(1~7)对肝癌细胞(HepG2)、结肠腺癌细胞(RKO)、卵巢腺癌细胞(OVCAR-3)共3种肿瘤细胞的体外抗肿瘤活性。 实验方法:取处于对数生长期的贴壁实验用肿瘤细胞按一定的细胞量(约含10000个肿瘤细胞,含10%胎牛血清的RMPI1640培养基作为单个细胞悬液)接种于96孔培养板中,每孔100 μL,再将其置于含5%CO2及100%湿度的37℃恒温箱中培养24 h。 将待测不同浓度的化合物溶液微滤除菌(0.22 μm),再分别稀释为10、5、2.5、1和0.5 μmol/L共5种浓度加入给药孔,每组设5个平行孔,同时做空白对照组孔(100 μL RMPI1640)。 将96孔板放回上述培养箱温育24 h后,每孔加入10 μL MTT,温育4 h,吸出上清液,向每孔中加入100 μL DMSO,室温下振摇,在酶标仪下用490 nm波长进行检测各孔OD值,并用以下公式计算细胞生长抑制率:
以化合物浓度对数值对抑制率作线性回归,得直线方程,从中求出半数抑制浓度IC50值。
化合物1~7是由藤黄酸C-30通过酰胺键或酯键分别接上具有不同长度碳链的腈基官能团所得的产物,由于反应物各不相同, 故采用不同的合成方法。 在化合物1、2的合成中,首先用DCC/HOSU体系,生成GA-HOSU活化酯中间体,活化酯中间体与3-氨基丙腈反应生成化合物2的过程较为顺利,但与氨基乙腈盐酸盐反应时加入有机碱三乙胺或DMAP均没有产物生成,后来采用先用碳酸铯中和氨基乙腈盐酸盐后再与活化酯中间体反应的方法,以53%的收率得到了目标产物1。
化合物5~7是由藤黄酸与相应的醇生成的酯。 最初,我们参考文献[20]的方法,选择1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDCI)和4-二甲氨基吡啶(DMAP)作为偶联催化剂制备酯,但反应的结果不理想,主要问题是反应的副产物较多,在薄层层析(TLC)检测中,发现多个与目标产物相近的杂质点,影响了产物的分离提纯。 后来,我们采用光延反应,以三苯基磷(PPh3)和偶氮二甲酸二乙酯(DEAD)为催化剂合成酯,结果发现效果较为理想,其优点一是反应的副产物少,并且在TLC中均与目标产物距离较远,容易分离提纯;二是反应只需在室温就可进行,反应条件温和。 该反应大部分化合物的产率都达到60%左右。
采用 MTT法测试了目标化合物1~7对人肝癌细胞HepG2、结肠腺癌细胞RKO、卵巢腺癌细胞OVCAR-3等3种肿瘤细胞的体外细胞增殖抑制活性,阳性对照物为藤黄酸。 实验结果见表1。
| 表1 不同化合物对3种肿瘤细胞的半数抑制浓度[IC50(μmol·L-1)] Table 1 The half inhibitory concentrations of three tumor cells treated with different compounds |
结果显示:1) 在C-30位通过酰胺键或酯键引入氰基的化合物2、3、4、6、7的抗肿瘤活优于GA。 其中氰基与酯键相隔2个碳原子的化合物4活性最强,对OVCAR-3癌细胞抗肿瘤活性大约是GA的4.5倍(IC50为1.41 μmol/L),同时对另外两种癌细胞也表现出不错的抗肿瘤活性;而相隔一个碳的化合物1活性最弱,说明氰基与酯键或酰胺键之间的碳链长度对化合物的活性有较大的影响,通过适当长度的碳链连接,有利于提高目标化合物的抗肿瘤活性。 2)含芳香氰基的藤黄酸衍生物也具有较好的抗肿瘤活性,化合物6对HepG2癌细胞抗肿瘤活性大约是GA的4倍(IC50为1.30 μmol/L) ,而且从体内代谢稳定性考虑,芳香氰基优于烷氰基[15]。 3)比较含对氰基苯基的化合物5与6,前者苯基与连接臂通过醚键连接,后者通过酰胺键连接,对人肝癌细胞HepG-2,后者的活性是前者的3.2倍;对卵巢腺癌细胞OVCAR-3,后者的活性是前者的2.4倍,说明芳基连接键种类对抗肿瘤活性也有较大的影响,酰胺键连接比醚键连接较为有利。
用紫外分光光度法测定了活性较好的化合物4、6及对照品GA在水中的溶解度,测得的溶解度分别为21.5、24.2和3.9 mg/L,与藤黄酸相比,化合物4和6的水溶性有了较大的提高。
以多靶点抗肿瘤天然产物藤黄酸为先导化合物,设计合成了7个藤黄酸氰基衍生物,其中6个为新化合物。 初步的体外抗肿瘤活性测试结果表明,化合物2、3、4、6、7表现出优于藤黄酸的抗肿瘤活性,能有效地抑制肝癌细胞(HepG2)、结肠腺癌细胞(RKO)、卵巢腺癌细胞(OVCAR-3)等3种肿瘤细胞的增殖,其中化合物4和6的活性最强,并且水溶性也有了较大的提高,可以作为抗肿瘤的候选化合物作进一步的研究。 藤黄酸衍生物的合成研究是近年来抗肿瘤药物研究的热点之一,本研究结果对藤黄酸衍生物的进一步优化设计具有一定的指导意义。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|