
ZHANG Guangchun, QIU Jian, XING Haiping, et al. Influence of Polyethylene- g-polystyrene on the Mechanical Properties and Foaming Behavior of Linear Low Density Polyethylene/Polystyrene Blends [J]. Chinese Journal of Applied Chemistry, 33(7): 756-765


共同通讯联系人:邢海平,助理研究员; Tel:0431-86262683; E-mail:hpxing@ciac.ac.cn; 研究方向:高分子改性与发泡材料
以通过开环易位聚合、加氢反应和原子转移自由基聚合技术结合制备的聚乙烯- g-聚苯乙烯(PE- g-PS)作为增容剂,研究了加入不同PS支链长度的PE- g-PS对于线性低密度聚乙烯/聚苯乙烯(LLDPE/PS)共混物的机械性能和发泡行为的影响。 以典型组成 m(LLDPE): m(PS)=70:30共混物为例,考察了PE- g-PS对共混物拉伸性能的影响。 相对于二元共混物,增容剂的加入使得断裂伸长率、拉伸强度和屈服强度皆提高,且含长PS支链的增容体系提高更明显。 采用超临界CO2釜式发泡工艺,考察了PE- g-PS中PS支链长度对共混物发泡行为的影响。 结果表明,相对于短PS支链体系,加入PE- g-PS1.59k(PS相对分子质量为1590)后的泡孔结构更加均一,完全没有“缝隙”形貌的出现。 当发泡温度降至80 ℃时,即使存在LLDPE发泡空间限制作用(LLDPE无法发泡),加入支链长度更长的PE- g-PS1.59k后泡孔分布也更加均一。
Co-corresponding author:XING Haiping, assistant professor; Tel:0431-86262683; E-mail:hpxing@ciac.ac.cn; Research interests:modification of polymer and foam materials
Polyethylene- g-polystyrene(PE- g-PS) copolymers, which were synthesized by the combination of ring-opening metathesis polymerization(ROMP), hydrogenation and atom transfer radical polymerization(ATRP) method, were utilized to compatibilize linear low density polyethylene/polystyrene(LLDPE/PS) blends. Using the LLDPE/PS blend in mass ratio of 70:30 as example, the effect of introduction of PE- g-PS on the mechanical properties was investigated. Compared to binary blends, elongation at break, tensile strength and yield strength are improved when the PE- g-PS is introduced. Utilizing PE- g-PS0.34k(the subscript 0.34k represents a relative molecular mass of PS is 340) and PE- g-PS1.59k as compatibilizers, the influence of PS branch length on foaming behavior of LLDPE/PS blends was investigated in a batch foaming process with supercritical carbon dioxide as physical foaming agent. Compared to the case containing PE- g-PS0.34k, the cellular structure of the foamed blends becomes much more uniform and no large gap appeares with the addition of PE- g-PS1.59k. When the foaming temperature is lowered to 80 ℃, confined effect of LLDPE solid around exists, much more uniform cellular structure also appears as PE- g-PS1.59k is introduced.





随着尖端技术、工农业生产和人们生活水平的不断提高,对材料性能提出了越来越高的要求,往往单一材料的性能已经无法满足使用要求,更多的需要多种性能并存。 对于固体材料或者相应的泡沫材料来说,我们更期待所制备的产品具备多种性能,能满足更高的要求[1]。 对于聚合物材料来说,比较简单的方法是将两种或者两种以上聚合物进行混合并用,在性能上取长补短,不仅具备材料成本的优势,还能在性能上超越单一材料所具备的性能[2]。 从热力学上来说,大多数聚合物之间是部分相容或者不相容的,无论物理混合还是混合后发泡过程,两相界面的调控是关键。 通常,需要通过添加增容剂来提高相界面间的相互作用,降低界面张力[3],最常用的增容剂是嵌段共聚物和接枝共聚物[4,5]。
众所周知,聚乙烯(PE)、聚苯乙烯(PS)是两种通用型的商业化高分子,且广泛用于生产和生活之中。 PE/PS共混物体系是典型的不相容体系,机械性能很差,一般需要添加增容剂(如PE- g-PS接枝共聚物)提高界面相互作用。 有报道通过电子束辐照[6,7]或者反应增容[8,9,10,11,12,13]的方法产生接枝共聚物来增容PE/PS共混物。 Díaz等[10]通过傅-克反应(Friedel-Crafts reaction)原位在PE/PS界面处产生接枝共聚物实现反应增容。 研究发现,即使催化剂质量分数低至0.3%,原位产生的共聚物也能产生显著的增容效果:界面黏附显著增强、分散相粒子尺寸减小。 与简单物理共混物相比,增容后的共混物机械性能得到提高。 无论采用反应增容还是电子束辐照的方法,反应过程皆很复杂,产物的微观结构无法表征清楚。 Chung等[14]通过茂金属催化共聚合与阴离子活性聚合技术的结合制备了PE- g-PS接枝共聚物。 这种制备方法能较好地控制共聚物组成和微观结构(接枝密度、接枝长度),PE- g-PS对高密度聚乙烯(HDPE)/PS共混物也表现出良好的增容效果。
要充分发挥不相容共混物泡沫的综合性能,共混物泡沫结构的控制是关键。 在前期发表的工作[15]中,我们研究了PE- g-PS的增容作用与线性低密度聚乙烯(LLDPE)/PS的组成(相态结构)对共混物发泡行为的协同影响。 研究发现,当LLDPE/PS二元共混物相结构为双连续相结构时,未增容共混物表现出很弱的发泡能力。 加入增容剂PE- g-PS后,LLDPE/PS/PE- g-PS的发泡能力提高。 未增容共混物在发泡成核时,双连续相结构的界面区(弱的界面黏附)可以为溶解的气体提供从基体扩散到周围环境中的通道,因此表现出很弱的发泡能力。 而加入增容剂后,溶解的CO2扩散到周围气氛中的通道由于界面区粘附增强而被“锁住”,因此发泡能力提高。 另一方面,LLDPE/PS二元共混物相结构为海岛相结构时,由于相界面区相互之间是分散且孤立的,增容剂的加入与否皆不影响CO2的扩散行为,故加入发泡剂后其发泡能力不发生显著改变。 同时,我们也发现,当增容剂添加含量固定时,提高增容剂PS支链长度,增容效果增强:两相界面黏附更强,分散相尺寸进一步减小。 根据我们前期的工作以及前人的工作,关于结构明确的增容剂支链长度等参数对增容后共混物发泡行为的影响的报道非常少。 因此,研究作为增容剂的接枝共聚物中支链长度对共混物发泡行为的影响以及如何优化界面行为来控制泡孔结构显得尤为重要。
在本工作中,通过开环易位聚合、加氢反应和原子转移自由基聚合技术结合制备了结构明确的PE- g-PS接枝共聚物,将其作为LLDPE/PS共混物的增容剂。 研究了具有不同PS支链长度的PE- g-PS对典型组成比 m(LLDPE): m(PS)=70:30共混物的机械性能以及发泡行为的作用。
线性低密度聚乙烯(LLDPE,DFDA-7042,熔体流动速率(MFR)=2.0 g/(10 min)(ISO1133,190 ℃,2.16 kg), Mw=1.41×105,多分散指数 Mw/ Mn(PDI)=3.3)购于茂名石化公司;聚苯乙烯(PS,Polyrex® PG-383,MFR=3.0 g/10 min(ISO1133,200 ℃,5 kg), Mw=3.72×105,PDI=1.7)购于镇江奇美化工有限公司;聚乙烯- g-聚苯乙烯共聚物(PE- g-PS)合成的具体实验步骤、实验表征方法见参考文献[15]。
Instron 1211型电子拉力机(美国Instron公司);XL 30 ESEM FEG型扫描电子显微镜(美国FEI公司);D8 ADVANCE型X射线衍射仪(德国Bruker公司);Mettler Toledo Star system(DSCI)型示差扫描量热仪(美国Mettler Toledo公司);不锈钢高压釜(2 L,自制)。
1.2.1 聚合物共混物的制备 所有的共混物样品均是通过溶液混合的方法制备。 LLDPE和少量2,6-二叔丁基-4-甲基苯酚(BHT)溶于回流的甲苯溶液中,同时N2气保护以防止氧化。 将PE- g-PS和PS加入到上述的热溶液中,当以上混合物形成透明、均一的溶液后,用乙醇进行沉淀,过滤,将沉淀物真空干燥24 h。
1.2.2 发泡样品制备 采用不锈钢高压釜进行釜式发泡制备发泡样品。 在高压釜里装入需要发泡的样品颗粒,低压CO2吹扫高压釜3~4次,用超临界泵注入一定量的CO2至釜内。 停止超临界泵打压,高压釜升到设定的发泡温度,继续采用超临界泵打压至所需的发泡压力。 所有的样品在超临界CO2中饱和4 h,保证CO2的平衡吸附。 饱和完成后,快速泄压至大气压,打开高压釜,取出发泡样品作测试用。
聚合物的相对分子质量及其相对分子质量分布采用带有示差检测器的高温凝胶渗透色谱(PL-GPC 220)进行测试。 聚合物溶液的配制过程为:约20 mg聚合物在150 ℃下溶于10 mL含有0.05%(质量分数)2,6-二叔丁基-4-甲基苯酚(BHT)抗氧剂的1,2,4-三氯苯溶剂中。 测试温度为150 ℃,流速为1 mL/min。
所有样品的熔点( Tm)、结晶温度( Tc)由DSC测定,N2气气氛,扫描温度范围25~200 ℃,分为升温-降温-二次升温三次扫描,升温和降温速率均为10 ℃/min,第一次升温到200 ℃停留5 min,其余扫描后停留1 min。 结晶度是基于样品的熔融焓值计算的,完全结晶的PE晶体熔融焓为293 J/g[16]。
广角X射线衍射(WAXD)采用Bruker D8 ADVANCE衍射仪,放射源为Cu Kα射线( λ=0.154 nm),操作电压为40 kV,电流为40 mA,测试范围为10°~50°,扫描速率为2°/min。
拉伸实验中,拉伸速度为20 mm/min。拉伸样条的有效部分尺寸为20 mm(长)×4 mm(宽)×1 mm(厚),每个样品至少测试五个平行样条,最终结果取平均值。
共混物及其发泡样品的形貌采用XL 30 ESEM FEG扫描电子显微镜(SEM)进行观察。 样品用液氮进行脆断,SEM观察前喷金处理。
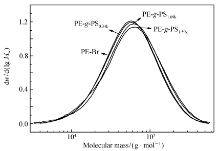
通过开环易位聚合、加氢反应和原子转移自由基聚合技术结合制备PE- g-PS接枝共聚物,将其作为LLDPE/PS共混物的增容剂。 经过核磁氢谱(1H NMR)[15]计算表明,支链密度为54条支链/10000主链碳原子,PS支链相对分子质量从340~1590变化。图1显示了线性聚合物PE-Br和PE- g-PS的高温GPC测试曲线,测得接枝共聚物的质均相对分子质量从74.8×103到82.2×103, Mw/ Mn=1.84~1.95。 分子结构表征结果总结于表1。
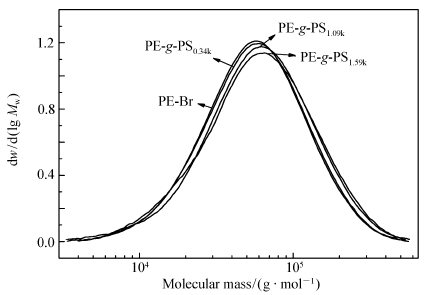 | 图1 PE-Br和PE- g-PS的GPC测试曲线Fig.1 GPC curves for the PE-Br and PE- g-PS |
{kind=link}
| 表1 PE-Br和PE- g-PS的分子结构参数总结 Table 1 Summary of the molecular characteristics of PE-Br and PE- g-PS copolymers |
图2给出了主链PE-Br和接枝共聚物PE- g-PS的DSC测试曲线,相应的DSC参数总结在表2中。 如表2所示,第二次升温曲线中,PE-Br和PE- g-PS熔点在117~119 ℃之间。 这表明,PS支链的引入并没有对主链PE-Br的熔点产生显著影响,文献中也有类似结果报道[17]。 我们认为,在结晶过程中,PS支链被排斥在晶片结构之外,并不影响PE晶体的完整性。 PE-Br的熔融焓为132.1 J/g,当引入PS支链后PE- g-PS熔融焓相比于PE-Br有所降低。 并且,随着PS接枝含量的增加,PE- g-PS的熔融焓呈下降的趋势。 事实上,接枝共聚物中只有主链PE有能力结晶,因此我们也考察了支链PS的引入对主链PE结晶度的影响。 当PE完全结晶熔融焓设定为293 J/g时,计算结果表明,PE- g-PS中PE部分与PE-Br有类似的结晶度,均为45%左右(表2)。 这说明PS支链的引入没有对PE的结晶度产生影响,而且随着支链长度的变化PE的结晶度也没有太大变化。 另一方面,降温曲线显示PE-Br和PE- g-PS的结晶温度在102~105 ℃之间。
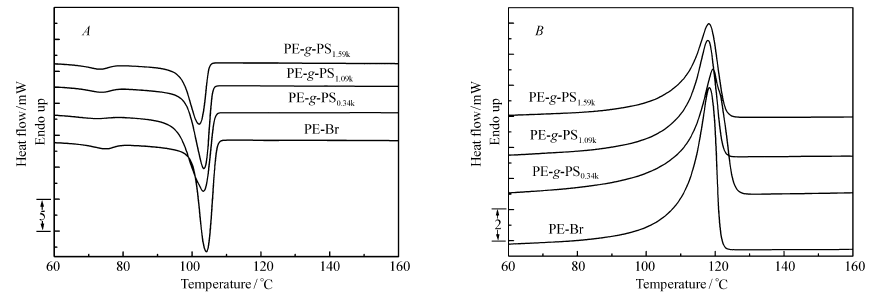 | 图2 PE-Br和PE- g-PS的降温( A)和第二次升温( B)DSC测试曲线Fig.2 DSC curves of PE-Br and PE- g-PS copolymers from cooling curves( A) and the second heating curves( B) |
{kind=link}
| 表2 PE-Br和PE- g-PS的DSC参数总结 Table 2 Summary of DSC parameters of PE-Br and PE- g-PS copolymers |
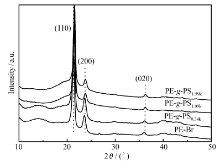
图3显示了PE-Br和PE- g-PS的WAXD图。 代表性的结晶衍射峰(110)、(200)、(020)出现在PE-Br中,同样这些结晶衍射峰也出现在PE- g-PS中,并且峰位也没有变化。 这表明支链PS的引入并没有影响主链PE的晶体结构。
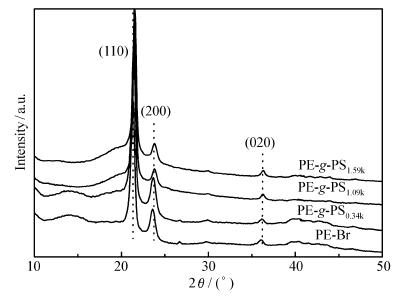 | 图3 PE-Br和PE- g-PS的WAXD图Fig.3 WAXD profiles of PE-Br and PE- g-PS copolymers |
{kind=link}
从前期工作[15]可知,PS分散相的平均尺寸随着增容剂浓度的提高逐渐减小,增容作用增强;并且,在同一添加含量下,随着接枝共聚物PS支链长度增加,PS分散相粒子尺寸也逐渐减小,增容效果增强。 本文研究了PE- g-PS的增容作用对LLDPE/PS共混物力学性能的影响。 以典型的共混物质量比 m(LLDPE): m(PS)=70:30为例,采用拉伸测试来对比增容前后共混物的机械性能变化。图4为加入增容剂PE- g-PS0.34k(下角标0.34k表示PS相对分子质量为340,下同)、PE- g-PS1.09k、PE- g-PS1.59k对共混物应力-应变曲线的影响。 拉伸性能参数(杨氏模量、屈服强度、拉伸强度以及断裂伸长率)总结于表3中。 一般来说,增容作用对断裂伸长率的影响十分敏感。 大的形变与界面粘附以及界面强度有很大的关系。 从表3可看出,相比于 m(LLDPE): m(PS)=70:30二元共混物,加入PE- g-PS1.09k、PE- g-PS1.59k后共混物的断裂伸长率增加,并且,随着增容剂浓度的提高,断裂伸长率显著提高。 以PE- g-PS1.09k为例,添加质量分数从1.0%增加到5.0%,断裂伸长率由113%增加到371%。 这主要是由界面粘附的增强以及分散相尺寸的减小所致。 然而,当PE- g-PS0.34k添加质量分数为1.0%和2.5%时,增容共混物显示低于二元共混物LLDPE/PS的断裂伸长率值。 当此增容剂添加质量分数提高至5.0%时,表现出高于二元 m(LLDPE): m(PS)=70:30共混物的断裂伸长率值。 这表明增容剂含量和PE- g-PS中PS支链长度对于共混物界面作用增强影响很大。 由表3可见,二元 m(LLDPE): m(PS)=70:30共混物显示很低的拉伸强度(6.7 MPa),这源于PS分散相与LLDPE基体弱的界面粘附(或界面相互作用)。 加入增容剂后,共混物的拉伸强度显著提高(约为 m(LLDPE): m(PS)=70:30共混物拉伸强度的两倍),这归因于界面粘附的增强。 拉伸过程中的屈服行为也能反映出增容作用的影响。 二元 m(LLDPE): m(PS)=70:30共混物的屈服强度为7.3 MPa,低于纯LLDPE的屈服强度值9.3 MPa。 而加入增容剂后,共混物表现出高的屈服强度(约为12 MPa),这是因为增容剂增强了界面作用所导致的。表3也列出了PE- g-PS加入后对共混物杨氏模量的影响。 由于杨氏模量数据是在较小拉伸变形过程中获得的(弹性变形),与界面的作用关系不大,主要依赖于共混物的聚集状态和组成。 与纯LLDPE相比,刚性PS材料的加入使得LLDPE/PS共混物以及增容后样品的杨氏模量提高。
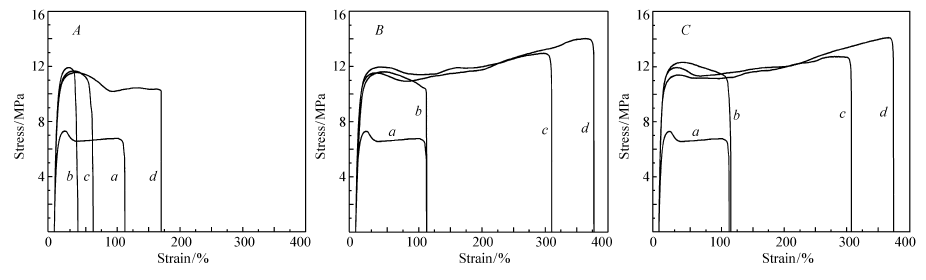 | 图4 m(LLDPE): m(PS)=70:30共混物以及加入PE- g-PS0.34k( A)、PE- g-PS1.09k( B)和PE- g-PS1.59k( C)增容剂后共混物的拉伸应力-应变曲线Fig.4 Stress-strain curves of m(LLDPE): m(PS)=70:30 blends and its compatibilized blends containing PE- g-PS0.34k( A), PE- g-PS1.09k( B), PE- g-PS1.59k( C) compatibilizers a. m(LLDPE): m(PS)=70:30; b. m(LLDPE): m(PS): m(PE- g-PS)=70:30:1.0; c. m(LLDPE): m(PS): m(PE- g-PS)=70:30:2.5; d. m(LLDPE): m(PS): m(PE- g-PS)=70:30:5.0 |
{kind=link}
| 表3 LLDPE、 m(LLDPE): m(PS)=70:30和增容后共混物的力学性能对比 Table 3 Comparison of mechanical properties of LLDPE, m(LLDPE): m(PS)=70:30 blend and its compatibilized blends |
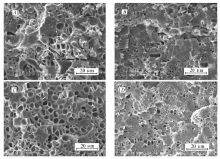
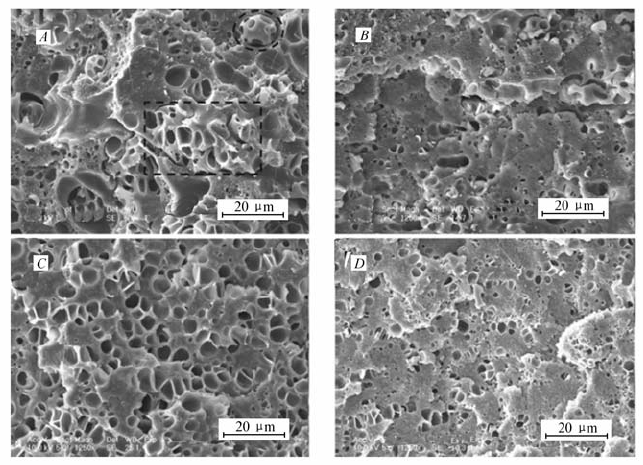
在前期工作中,我们发现PE- g-PS对LLDPE/PS共混物发泡行为的影响强烈依赖于共混物的组成(相态结构),并系统研究了增容作用和LLDPE/PS共混物的组成二者相互作用对共混物发泡行为的影响[15]。 然而,我们只研究了单一增容剂(PE- g-PS0.34k或者PE- g-PS1.59k)的加入量对发泡行为的影响,没有系统对比在增容剂添加含量相同时,PE- g-PS中PS支链长度变化对共混物发泡行为的作用。 因此,在此工作中,以PE- g-PS0.34k、PE- g-PS1.59k作为增容剂,发泡条件为发泡温度108 ℃,CO2饱和压力13.3 MPa,饱和时间4 h,系统研究了PS支链长度对LLDPE/PS共混物发泡形貌的影响。 从扫描电子显微镜结果(图5 A)可以看出,加入增容剂PE- g-PS0.34k后,共混物 m(LLDPE): m(PS): m(PE- g-PS0.34k)=70:30:1.0表现出良好的发泡能力。 然而,在当前发泡条件下,泡孔结构中有些区域出现很大的“缝隙”,如图5 A虚线圆圈所示。 对于增容共混物 m(LLDPE): m(PS): m(PE- g-PS0.34k)=50:50:1.0,泡孔形貌中同样出现结构上的不均一(图5 B虚线圆圈所示)。 当加入PS支链长度更长的增容剂PE- g-PS1.59k后,如图5 D所示, m(LLDPE): m(PS): m(PE- g-PS1.59k)=70:30:1.0的泡孔形貌变得非常均匀,泡孔结构中完全没有“缝隙”的出现。 同样, m(LLDPE): m(PS): m(PE- g-PS1.59k)=50:50:1.0表现出类似的现象(图5 E)。
 | 图5 m(LLDPE): m(PS)=70:30、50:50、80:20共混物中加入PE- g-PS0.34k( A, B, C)、PE- g-PS1.59k( D, E, F)后的泡孔形貌SEM照片Fig.5 SEM images of foamed m(LLDPE): m(PS): m(PE- g-PS0.34k)=70:30:1.0( A), m(LLDPE): m(PS): m(PE- g-PS0.34k)=50:50:1.0( B), m(LLDPE): m(PS): m(PE- g-PS0.34k)=80:20:1.0( C), m(LLDPE): m(PS): m(PE- g-PS1.59k)=70:30:1.0( D), m(LLDPE): m(PS): m(PE- g-PS1.59k)=50:50:1.0( E) and m(LLDPE): m(PS): m(PE- g-PS1.59k)=80:20:1.0( F) samples |
{kind=link}
 | 图6 m(LLDPE): m(PS): m(PE- g-PS0.34k) =70:30:1.0( A)和 m(LLDPE): m(PS): m(PE- g-PS1.59k)=70:30:1.0( B)未发泡样品的SEM脆断面形貌图Fig.6 SEM images of unfoamed m(LLDPE): m(PS): m(PE- g-PS0.34k)=70:30:1.0( A) and m(LLDPE): m(PS): m(PE- g-PS1.59k)=70:30:1.0( B) samples |
{kind=link}
当共混物组成比固定时,加入支链长度更长的增容剂后,PS分散相尺寸变得更小,表现出更好的增容效果。 例如,在 m(LLDPE): m(PS)=70:30中加入增容剂PE- g-PS0.34k后,虽然脆断表面变得更加平整,但是仍然存在大尺寸不规则形状的PS粒子以及些许凹面形貌(图6 A),而加入PE- g-PS1.59k后脆断表面平整,无凹凸形貌,且PS粒子尺寸变小、粒子分布均匀,形状趋于球形(图6 B)。 这表明,相对于PE- g-PS0.34k,加入PE- g-PS1.59k后两相界面粘附更强,增容效果更好。 当饱和时间结束泄压时,两相界面处容易成核,基体中溶解的CO2快速向界面处聚集。 由于大尺寸不规则形状的分散相粒子周围界面面积更大,向此界面处扩散的CO2更多,同时这些区域的界面粘附没有那么强,在泡孔长大膨胀的过程中,界面处不同聚合物组分容易相互分离,故表现出较大的“缝隙”形貌。 相反,加入PE- g-PS1.59k后两相界面粘附力很强,泡孔长大过程中界面处的聚合物不会分离,故完全没有“缝隙”形貌的出现。 另一方面,当加入PE- g-PS0.34k后,未发泡增容共混物中PS分散相粒子尺寸分布极不均匀(不规则的PS大尺寸粒子和球形粒子并存),发泡过程中相界面诱导的成核点分布也不均匀,故发泡后样品的泡孔尺寸分布也极不均匀,如图5 A所示。 相反,当加入PE- g-PS1.59k后,未发泡增容共混物中分散相粒子形状趋于球形(呈海岛结构)且在基体中分布均匀,因此在发泡过程中由相界面诱导的成核点分布也更加均匀,形成的泡孔尺寸分布自然也就更均匀,如图5 D所示。我们对发泡样品 m(LLDPE): m(PS): m(PE- g-PS0.34k)=70:30:1.0与 m(LLDPE): m(PS): m(PE- g-PS1.59k)=70:30:1.0用四氢呋喃(THF)溶剂进行了刻蚀,将其中PS组分选择性刻蚀掉(常温下刻蚀时间为24 h),刻蚀后的形貌如图7所示。 对于 m(LLDPE): m(PS): m(PE- g-PS0.34k)=70:30:1.0发泡样品,刻蚀后泡孔结构出现大量的凹坑;与此相反,发泡样品 m(LLDPE): m(PS): m(PE- g-PS1.59k)=70:30:1.0在刻蚀后泡孔结构整体上则十分完整,没有凹坑形貌出现。图5 A与图7 A对比可知,大量被刻蚀掉的区域主要位于“缝隙”周围的区域。 如前分析,“缝隙”主要因大尺寸不规则形状的PS区与PE区在泡孔长大过程中相互分离所致,那么在刻蚀过程中裂缝周围PS区域被刻蚀掉,也就相应的留下大量凹坑形貌。 相反地,由于 m(LLDPE): m(PS): m(PE- g-PS1.59k)=70:30:1.0共混物中PS粒子分散均匀,尺寸较小,且两相界面粘附很强,在泡孔长大膨胀过程中,PE、PS两相被同时挤压至泡孔壁。 与图5 D完整的闭孔泡孔形貌相比,刻蚀后的泡孔结构出现一些开孔结构,同时还存在一些丝状结构(图7 B虚线圆圈所示),这主要是泡孔壁中PS部分被大量刻蚀所留下的PE部分。 从刻蚀结果也进一步说明,PS支链长度变化对增容共混物泡孔结构的不同作用。 我们也对具有海岛相结构的 m(LLDPE): m(PS)=80:20增容共混物的发泡行为受PS支链长度的影响进行了研究,相应的泡孔形貌如图5 C与5 F所示。 类似地,当PS支链长度较短时, m(LLDPE): m(PS): m(PE- g-PS0.34k)=80:20:1.0共混物泡孔形貌中仍有少量“缝隙”出现,当增容剂PS长度增加至1.59k时,LLDPE/PS/PE- g-PS1.59k共混物的泡孔形貌变得非常完整,且泡孔分布均匀。 这些现象产生的原因仍是不同PS支链长度的增容剂对界面行为的调控效果不同所致。
 | 图7 发泡样品 m(LLDPE): m(PS): m(PE- g-PS0.34k)=70:30:1.0( A)与 m(LLDPE): m(PS): m(PE- g-PS1.59k)=70:30:1.0( B)刻蚀后形貌SEM照片Fig.7 SEM images of the etched morphology of foamed m(LLDPE): m(PS): m(PE- g-PS0.34k)=70:30:1.0( A) and m(LLDPE): m(PS): m(PE- g-PS1.59k)=70:30:1.0( B) samples |
{kind=link}
当保持其它发泡条件不变,发泡温度降至80 ℃时,我们研究了在相对低的发泡温度下,PS支链长度变化对增容后共混物发泡行为的影响,典型的发泡形貌如图8所示。 如图8 A所示, m(LLDPE): m(PS): m(PE- g-PS0.34k)=70:30:1.0发泡样品中存在大量的未发泡区域,发泡区域主要包括一些分布于分散相上类似半球形的泡孔(图8 A虚线圆圈所示)以及大面积聚集的泡孔(图8 A虚线矩形区域)两区域。 相反地,加入PS支链长度较长的增容剂后, m(LLDPE): m(PS): m(PE- g-PS1.59k)=70:30:1.0发泡样品的泡孔结构分布相对均匀,如图8 C所示。 在当前的发泡条件下,由于发泡温度过低,LLDPE是处于完全不能发泡的状态而PS则是完全发泡的泡孔形貌。 如前所述,当加入增容剂PE- g-PS0.34k后, m(LLDPE): m(PS): m(PE- g-PS0.34k)=70:30:1.0未发泡样品中较小的PS球形粒子与大尺寸不规则PS粒子形貌共存。 由于加入增容剂后两相界面张力降低,泄压时趋向于向两相界面处成核,体系中溶解的CO2向相界面处扩散聚集,泡孔长大过程中LLDPE固体区域表现出的空间限制作用,使得泡孔向PS区域膨胀延伸,故在PS球形粒子表面形成类似半球形的泡孔结构。 相对地,在大面积的不规则PS区域则出现泡孔聚集的形貌。 相反地,加入PS支链长度更长的PE- g-PS1.59k后, m(LLDPE): m(PS): m(PE- g-PS1.59k)=70:30:1.0样品中PS分散相尺寸减小且分布均匀,那么泄压成核时两相界面处诱发的成核点则更加均匀,故形成泡孔分布均匀的泡孔形貌。 同时,还注意到,由于 m(LLDPE): m(PS): m(PE- g-PS0.34k)=70:30:1.0样品界面处粘附力不是很强,故泡孔长大过程中出现PS泡孔区域与LLDPE剥离,两相界面处空隙增大的现象。 对于 m(LLDPE): m(PS): m(PE- g-PS1.59k)=70:30:1.0样品来说,两相界面粘附力更强,故泡孔结构中并未出现界面处相互剥离的现象。 另一方面,相对于 m(LLDPE): m(PS): m(PE- g-PS0.34k)=80:20:1.0样品来说, m(LLDPE): m(PS): m(PE- g-PS1.59k)=80:20:1.0发泡样品泡孔区域增加,泡孔结构均匀的原因也是由于不同PS支链长度对两相界面行为不同调控结果所致。
 | 图8 m(LLDPE): m(PS)=70:30、80:20加入PE- g-PS0.34k( A, B)、PE- g-PS1.59k( C, D)后的泡孔形貌SEM照片Fig.8 SEM images of foamed m(LLDPE): m(PS): m(PE- g-PS0.34k)=70:30:1.0( A), m(LLDPE): m(PS): m(PE- g-PS0.34k)=80:20:1.0( B), m(LLDPE): m(PS): m(PE- g-PS1.59k)=70:30:1.0( C), m(LLDPE): m(PS): m(PE- g-PS1.59k)=80:20:1.0( D) samples |
{kind=link}
通过开环易位聚合、加氢反应和原子转移自由基聚合技术结合制备PE- g-PS接枝共聚物,PS支链的引入并没有对主链PE的熔点和晶体结构产生影响。 以典型的质量比 m(LLDPE): m(PS)=70:30共混物为例,研究了PE- g-PS中PS支链长度变化对LLDPE/PS共混物机械性能的影响。 加入增容剂PE- g-PS1.09k、PE- g-PS1.59k后,共混物的断裂伸长率、拉伸强度、屈服强度皆提高。 这些现象归因于良好的界面粘附以及分散相尺寸的减小。 PS支链长度对LLDPE/PS共混物发泡形貌的影响结果表明,支链长度增加时,泡孔结构更加均匀,不会出现大的“缝隙”(发泡温度108 ℃)泡孔形貌。 这主要归因于不同PS支链长度对两相界面行为不同调控结果导致成核、泡孔长大过程不同。 当两相组成质量比为80:20或者降低发泡温度至80 ℃,PS支链长度引起泡孔形貌的差异也是由上述原因所引起的。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|