
ZHOU Wenjun, DENG Jiaying. Research Progress of Iron-catalyzed Cross-coupling Reactions[J]. Chinese Journal of Applied Chemistry, 33(3): 245-266


过渡金属催化的交叉偶联反应具有反应条件温和、操作简单、底物范围广等特点而备受关注,其中,铁在地球上存储丰富、廉价、无毒、对环境友好,商品化的铁盐和含铁化合物种类繁多,是一种可供选择的、绿色的、较为理想的催化剂。 近年来铁作为催化剂应用于交叉偶联反应的相关报道逐渐增多,本文根据铁催化剂的不同种类,综述了近年来铁盐催化交叉偶联反应的研究进展,期望为相关的研究工作提供参考。
Transition metal-catalyzed cross-coupling reaction which has advantages of simple operation, mild reaction conditions, and broad scope of substrates is one of the most efficient methods for the construction of carbon-carbon bonds. Iron is one of the potential green catalysts as it is one of the most abundant, inexpensive metals on earth, and environmentally friendly. Iron salts and iron-containing compounds are widely commercially available. Recently, more and more studies report the use of iron as catalyst in cross-coupling reactions. This paper reviews the recent advances in iron-catalyzed cross-coupling reactions based on different raw materials and we hope to provide some inspiration for relative research.


































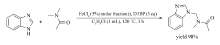






交叉偶联反应是指有机亲电试剂与有机亲核试剂在过渡金属催化下构建C—X(X=C、O、N、S等)键的反应[1,2,3,4,5,6](Scheme 1)。 过去的研究常用卤化物作亲电试剂[7],近几年的报道显示,用廉价易得的酚的衍生物也可做亲电试剂[8,9,10,11]。 多数交叉偶联反应具有反应选择性好、催化剂的催化效率高、反应所需条件温和、反应底物来源广泛的特点,该类型的反应不仅在有机合成化学以及催化化学领域成为研究热点之一,同时在高分子材料、医药生产、液晶材料以及功能材料的合成领域得到广泛的应用[12,13]。
 | Scheme 1 Cross-coupling reaction |
由于钯、镍和铜具有反应底物适用范围广,催化活性高,所需的反应条件温和等特点,因此选择钯、镍和铜作为交叉偶联反应最常见的催化剂。 但钯和镍面临有毒、体系昂贵、不易回收等问题,造成钯和镍催化剂在交叉偶联反应中不能广泛的应用[14],铜催化剂虽然避免了价格昂贵,有毒等特点,但对活性较低的氯代芳烃的催化效果很差[15]。 铁是自然界中较为丰富的金属,它具有无毒无害、对环境友好、廉价且存在大量的市售铁盐和配合物等优点,因此是较为理想的催化剂[16,17]。 事实上,在过去的10年,使用易于处理的铁盐作催化剂,在有机合成中构建碳碳键已取得一定的成功[18,19],在碳碳键、碳氮键、碳硫键形成等领域应用的报道逐渐增多。 本文主要介绍了近几年铁作为催化剂催化交叉偶联反应的研究进展。
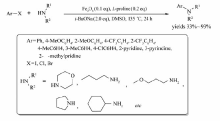
作为一种重要的铁盐,FeCl3价格低廉,作为催化剂被广泛应用到构建C—C键的偶联反应之中。 Bolm等[20]报道了FeCl3催化末端炔烃与芳基碘的偶联反应(Scheme 2)。 体系以甲苯为溶剂, N, N'-二甲基乙二胺(DMEDA)为配体,Cs2CO3为缚酸剂,在132 ℃条件下反应72 h,以15%~99.9%的产率获取目标产物。
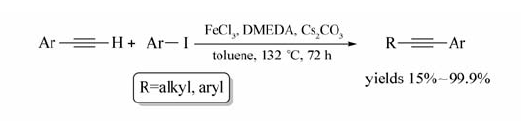 | Scheme 2 FeCl3 catalyzed cross-coupling reaction between terminal alkynes and aryl iodides[20] |
共轭烯炔是重要的生物活性分子,是某些药物中间体、电子材料中重要的结构单元,近几年的研究表明,过渡金属铁是合成烯炔的重要催化剂之一[21]。 2008年,Nakamura等[22]提出了FeCl3催化末端炔烃与烯基溴或烯基三氟甲磺酸酯合成烯炔的反应(Scheme 3)。 由末端炔烃在FeCl3/LiBr的催化下生成炔基格氏试剂,然后在60 ℃条件下,与烯基溴化物或烯基三氟甲磺酸酯反应形成相应的目标产物。
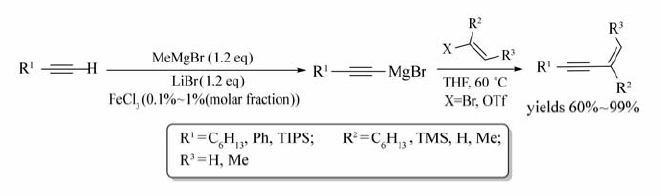 | Scheme 3 FeCl3 catalyzed cross-coupling reaction between terminal alkynes and aryl bromides or aryl triflates[21] |
2009年,Xie等[23]通过FeCl3催化,实现了末端炔烃与烯基碘化物合成共轭烯炔衍生物的反应(Scheme 4)。 该反应条件温和,对环境友好,获得的目标产物产率高(52%~90%)。
 | Scheme 4 FeCl3 catalyzed cross-coupling reaction between terminal alkynes and aryl iodides[23] |
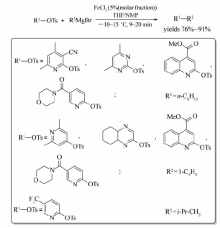
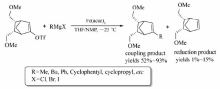
-10~15 ℃的条件下,以THF/NMP为溶剂,杂环磺酸酯在FeCl3催化下能与烷基格氏试剂发生交叉偶联反应[24](Scheme 5)。 对甲苯磺酰氧基(OTs)被格氏试剂的烃基取代,反应只需9~20 min即可完成,目标产物产率可达76%~91%。 铁的其他化合物,如FeCl2、Fe(acac)3对该反应均有一定的催化效果,相同反应条件下,杂环磷酸酯、杂环胺基磺酸酯与烷基格氏试剂的偶联反应也能顺利进行。
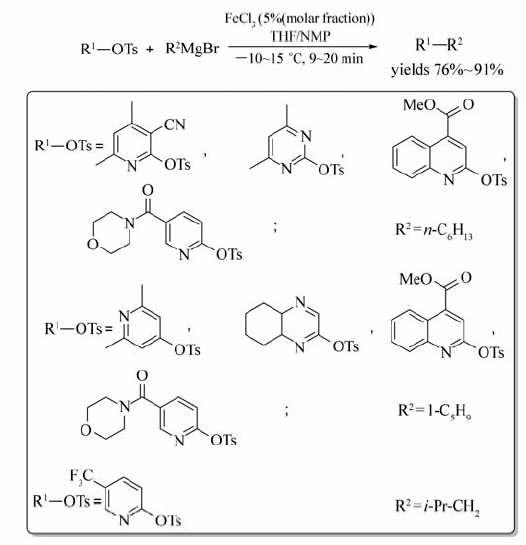 | Scheme 5 FeCl3 catalyzed cross-coupling reaction between heterocyclic sulfonate and alkyl Grignard reagent[24] |
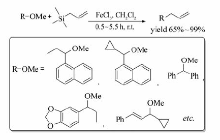
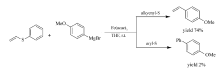
FeCl3也可催化苄/烯丙基醚与烯丙基硅烷发生交叉偶联反应[25](Scheme 6),该法不仅可顺利地实现C( sp3)—O键断裂和C( sp3)—C( sp3)键的构建,也能高效地引入C=C键。 含C=C键、环丙基以及芳香甲氧基的醚均可作为反应底物,以高达99%的产率获得相应的目标产物。
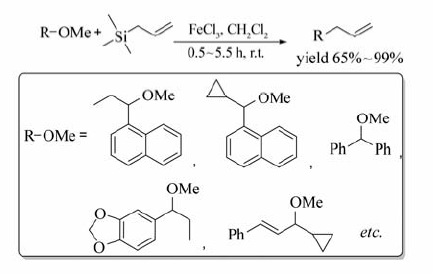 | Scheme 6 Cross-coupling reaction between benzyl/isopropyl ether and allylsilane[25] |
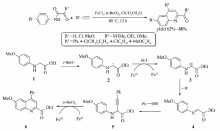
Liu等[26]报道了FeCl3催化甘氨酸衍生物合成喹啉类衍生物的交叉偶联脱氢反应(CDC)(Scheme 7)。 80 ℃条件下,以1,2-二氯乙烷为溶剂,在FeCl3/( t-BuO)2作用下,甘氨酸衍生物与末端炔烃能够顺利地发生反应,以62%~88%的产率获取目标产物。 进一步研究发现含有酯基的炔烃并不能发生该反应,当某些甘氨酸衍生物做底物(R1=MeO, R2=MeO, OEt时),改用CHCl3为溶剂,反应只需在60 ℃下进行1h便可获取相应的产物。 该反应为喹啉及其衍生物的合成提供了一个简捷而经济的方法。
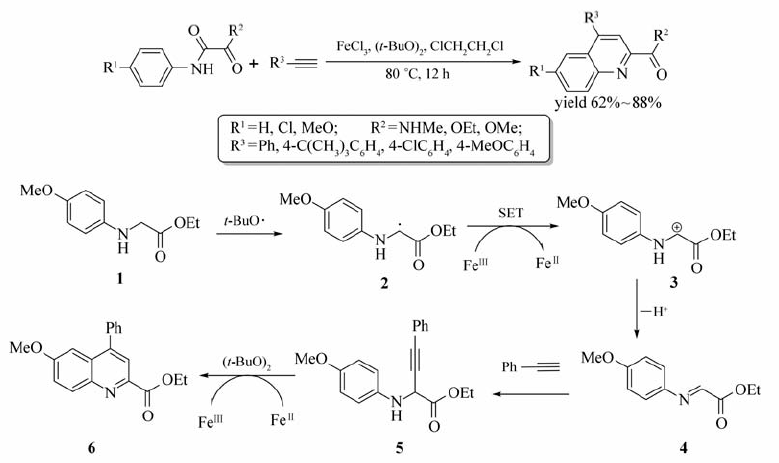 | Scheme 7 Dehydrogenation cross-coupling between terminal alkynes and glycine derivatives and possible mechanism[26] |
通过研究,Liu等[26]认为该反应经历了一个自由基中间体历程,首先在铁盐催化下( t-BuO)2发生均裂产生叔丁氧基自由基( t-BuO·),化合物1中的 α氢与该自由基作用形成自由基2,此时,在铁盐作用下发生一个单电子转移过程(SET)形成正离子3,之后发生 α氢消除而形成亚胺4,炔烃与亚胺4发生亲核加成得到中间体化合物5,最后在铁盐的催化下分子内发生类似Friedel-Crafts反应而形成最终目标产物。
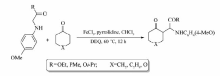
此外,甘氨酸衍生物在FeCl3/2,3-二氯-5,6-二氰基-1,4-苯醌(DDQ)催化氧化下,也能与环酮在发生交叉偶联脱氢反应[26](Scheme 8)。 根据文献报道,环状酮类衍生物能顺利发生该反应,产率可达63%~83%。 遗憾的是,直链酮(如丙酮和苯乙酮)并不能发生。 该反应条件温和,产物的产率较好,且化学选择性好,为氨基酸衍生物引入新的官能团提供了有效的方法。
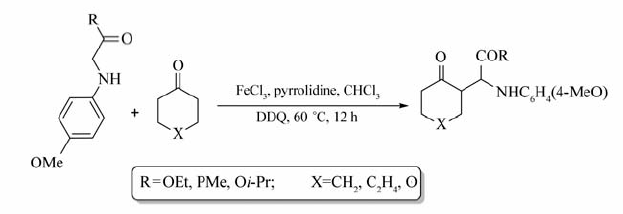 | Scheme 8 Dehydrogenation cross-coupling between cyclic ketones and glycine derivatives[26] |
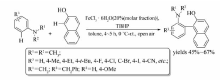
一定条件下,FeCl3·6H2O催化 N, N-二甲基苯胺与2-萘酚也能发生交叉偶联脱氢反应[27](Scheme 9)。 研究显示O2、H2O2、DDQ、 N-溴代丁二酰亚胺(NBS)等做氧化剂时几乎没有目标产物产生。 改用叔丁基过氧化氢(TBHP)作催化剂时,反应24 h,目标产物产率可达67%。 该反应条件温和,无需隔绝空气和水,无需单独引入或脱除官能团,且区域选择性强,是合成具有不同官能团的芳基化合物的有效的方法。
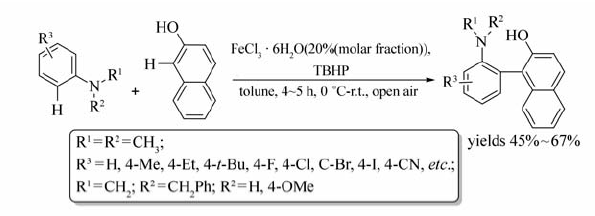 | Scheme 9 Cross-coupling reaction between N, N-dimethylaniline derivatives and 2-naphthol[27] |
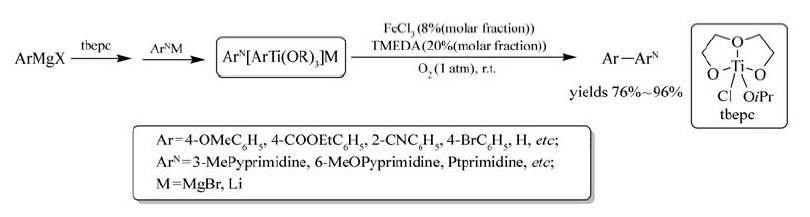
Liu等[28]采用 N, N, N', N'-四甲基乙二胺(TMEDA)为配体,实现了FeCl3/O2催化氧化 N-杂环化合物与芳基格氏试剂或芳基锂的偶联反应(Scheme 10)。 该反应成功的关键是反应起始时加入钛酸与芳基卤化镁转化为相应的ArTi(OR)3,ArTi(OR)3再与相应的 N-杂环芳基化合物反应形成中间体ArN[OR3]M,最后在TMEDA中,FeCl3/O2催化氧化形成目标产物Ar-ArN。 钛酸(tbepc)的加入不仅可以有效地提高目标产物的产率,还可避免格氏试剂或 N-杂环芳基化合物自身偶联反应的形成。 吡啶、2-苯基吡啶、喹啉、异喹啉、苯并噻唑、吡唑、嘌呤等化合物均可顺利发生该反应,并可获取良好产率的目标产物,而钯和镍催化体系不能实现吡啶衍生物参与的反应。
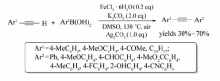
2009年,Daivd等[29]实现了FeCl3/KF催化芳基溴、碘与芳基硼酸的Suzuki偶联反应(Scheme 11)。 反应以乙醇为溶剂,在100 ℃条件下密封管内作用16~36 h,能高产率地形成联芳烃化合物(77%~99%)。 刘岩等[30]发现,若改用FeCl2·4H2O作催化剂,K2CO3为碱,上述反应同样能高效地进行,且反应温度仅需80 ℃。 研究表明,含有氟原子、环烷基等取代基的芳基溴化物,以及含氟的芳基硼酸均可顺利进行该反应。 该方法操作简单,经济环保,不仅扩大了铁盐的应用范围,同时为联芳烃化合物的制备提供了新的方法。

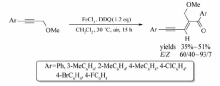
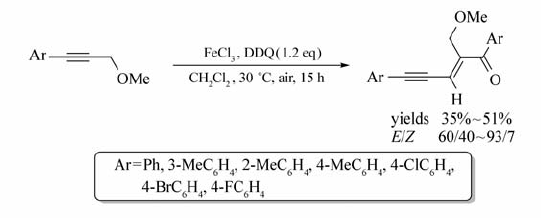
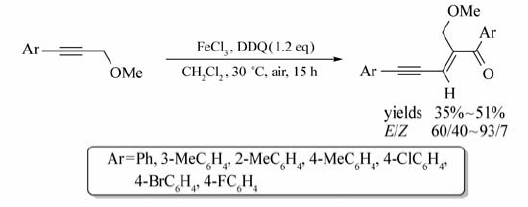
Xie等[31]报道了以FeCl2为催化剂,DDQ为氧化剂,芳基炔丙基醚与 β-二羰基化物的氧化偶联反应(Scheme 12)。 该体系以CH2Cl2为溶剂,在15 ℃条件下作用15 h,FeCl2能催化芳基炔丙基醚的C( sp3)—H键活化,并以中等产率获取目标产物(37%~64%)。 研究发现,以FeCl3作催化剂时,其它反应条件不变,芳基炔丙基酮也能发生自身偶联反应,合成相应的烯酮化合物。
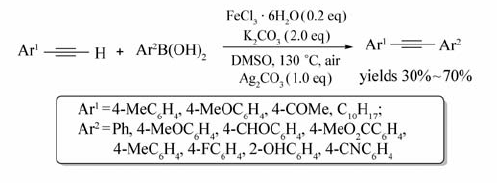
以DMSO为溶剂,K2CO3为碱,Ag2CO3为添加剂,FeCl3·6H2O为催化剂,末端芳炔与芳基硼酸能发生偶联反应生成相应的二芳基炔烃[32](Scheme 13)。 该反应无需N2气等惰性气体的保护,在130 ℃的条件下,反应24 h后,以30%~70%的产率获得相应的二芳基炔烃。 该反应适用范围广,芳环上的甲基、甲氧基、氰基、酯基、酰基等基团对反应无明显影响。 值得一提的是,Ag2CO3的加入可以有效避免末端炔烃自身偶联产物的形成。
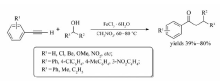
Liu等[33]报道了FeCl3·6H2O催化苄醇与烯烃的交叉偶联反应(Scheme 14)。 该反应的立体选择性强,产物最终仅形成 E型的取代烯烃,能实现该反应的底物达15例,其中合成目标产物的产率最高可达99%。 反应条件温和,原子经济性好,对环境友好,立体选择性高等优点,使得该反应有较广的应用前景。

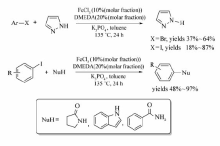
除了C—C键的构建,FeCl3还可用到C—N键的构建之中。 通过卤代烃参与偶联,Blom等[34]成功实现了 N-芳基化反应(Scheme 15),该方法与已知的钯或铜催化的 N-烷基化交叉偶联反应相比,提供了一种绿色、环保的FeCl3/DMEDA催化体系。
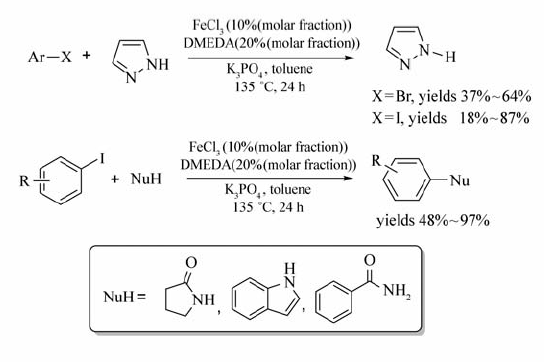
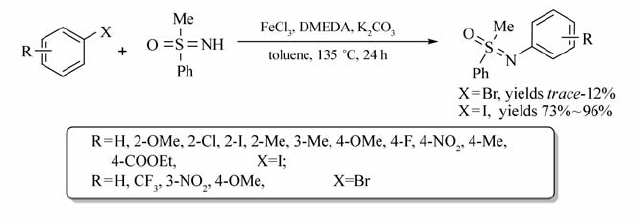
利用该催化体系,通过卤代芳烃与亚磺酰亚胺的交叉偶联反应,可实现各种芳基磺酰胺类化合物的合成(Scheme 16)[35]。 反应中,碘代芳烃以较好的产率获得目标产物(73%~96%),溴代芳烃所得的产率很低(仅为12%)。 而氯代芳烃,根本不能发生该反应。
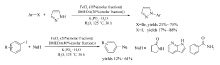
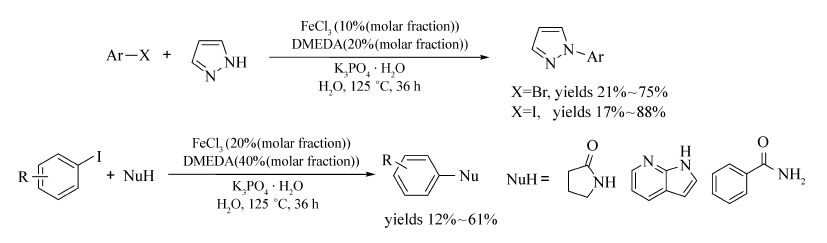
该催化体系也能实现水相中的 N-烷基化交叉偶联反应[36](Scheme 17),该方法避免了有机溶剂的使用,采用无毒无害、自然界广泛存在的水作为溶剂,使得该方法向着环境友好的绿色合成更加靠近。
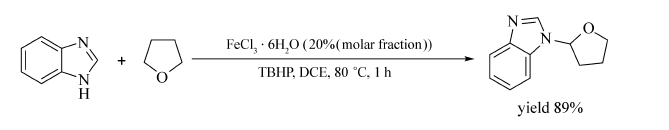
Pan等[37]通过醚类化合物 α位的C—H活化,实现了 N-取代苯并咪唑类衍生物的合成(Scheme 18)。 在ClCH2CH2Cl或EtOAc中,80 ℃条件下,苯并咪唑衍生物能与醚(环状醚或无环醚)发生偶联,以40%~91%的产率获取相应目标产物。 该反应条件温和,底物范围广,为 N-取代苯并咪唑类衍生物的合成提供了重要的方法。
Yao等[38]在甲苯中加入K2CO3,FeCl3·6H2O/DMEDA,在90 ℃条件下实现了吡咯烷酮与芳基炔溴的偶联反应,成功合成了芳基炔基酰胺类化合物(Scheme 19)。 该反应底物适用范围广,反应条件温和,操作简单,产率高(57%~90%),为芳基炔基酰胺的合成提供了有效的参考。
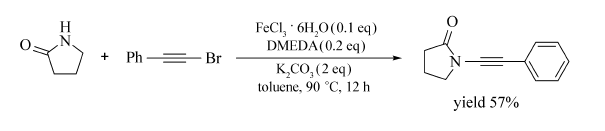 | Scheme 19 FeCl3·6H2O catalyzed cross-coupling reaction between 1-bromo-2-phenylacetylene and pyrrolidin-2-one[38] |
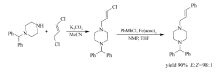
乙烯基溴化物或氯化物与咪唑的交叉偶联反应[39]也可通过铁盐催化实现(Scheme 20)。 研究显示, E-乙烯基溴化物作反应底物时, Z型产物占主导( Z>60%),而 E-乙烯基氯化物作底物,则是获取 E型的产物( E>50%)。 值得一提的是,当 E-乙烯基氯化物做底物,体系的反应温度超过120 ℃时,产物的产率有明显的提高,但 E型产物所占的比例将降低,特别是当反应时间超过24 h时,所得产物的产率和 E型异构体均有所降低;若体系加入10%摩尔分数的CuI,虽然产物产率降低,但获得的产物中 E型异构体达90%以上。
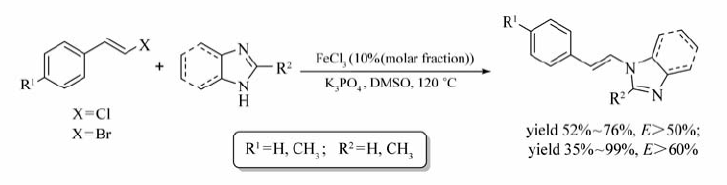
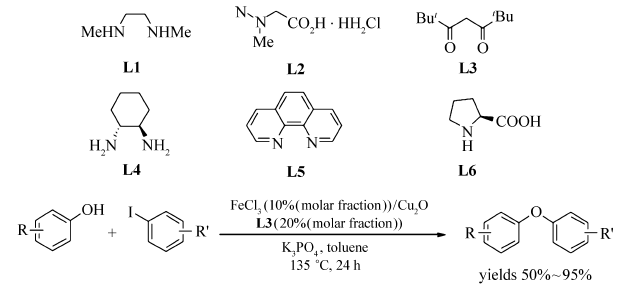
Bolm等[40]通过对配体(L1~L6)的筛选,实现了FeCl3催化的 O-芳基化反应(Scheme 21)。 反应选用Cu2O作为共催化剂,2,2,6,6-四甲基-3,5-庚二酮(TMHD)为配体,Cs2CO3为碱,DMF为溶剂,130 ℃条件下顺利实现酚类化合物与碘代芳烃的 O-烷基化交叉偶联反应。
Yang等[41]成功实现了FeCl3/1,10-phenanthroline催化邻碘苯酚与末端芳炔的偶联反应,合成了系列2-芳基苯并呋喃化合物衍生物(Scheme 22)。 该反应以Cs2CO3为碱,在N2气氛围中,120~145 ℃的条件下反应36 h,以较好的产率(43%~85%)得到相应的目标产物。 研究还发现,邻碘苯酚与1-芳基-2-三甲基硅烷乙炔在相同条件下也能高产地合成2-芳基苯并呋喃。
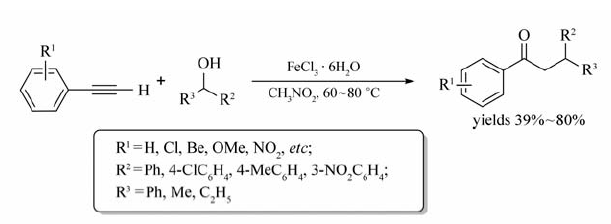
Jana等[42]以硝基甲烷为溶剂,成功实现了FeCl3·6H2O催化末端芳炔与苄醇的反应,生成取代芳酮类化合物(Scheme 23)。 研究表明,不同取代的末端芳炔均能与相应的苄醇反应,以39%~80%的产率获取芳基酮类化合物,研究表明富电子炔烃比缺电子或中性炔烃更易发生反应,相应产物的产率也更高。
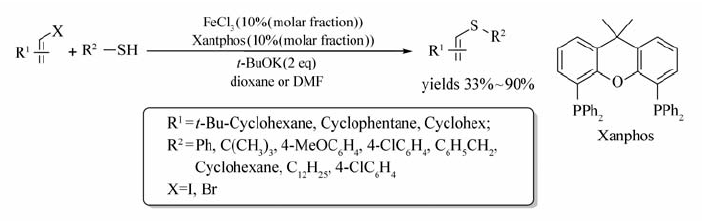
烯基卤化物与硫醇的偶联反应也可通过铁催化实现[43](Scheme 24)。 该体系以Xantphos为配体,DMF作溶剂, t-BuOK为碱,实现了硫氢键的活化。不同的烯基溴化物或碘化物均能与相应的硫醇反应,以33%~99%的产率获取目标产物。 该反应与传统硫醇的偶联反应相比,克服了对底物 β-溴苯乙烯, β-氯苯乙烯不反应的缺点,并能高产率地合成目标产物。
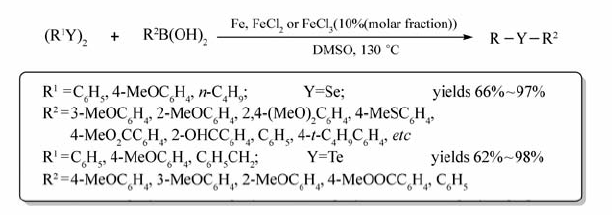
Wang等[44]实现了铁单质、FeCl2或FeCl3催化芳基硼酸与二硒醚或二碲醚构建C—Se键或C—Te键的交叉偶联反应(Scheme 25)。 通过对反应条件进行筛选发现,以DMSO为溶剂,在130 ℃的条件下可获取最高产率的目标产物。 各类取代芳基硼酸均可顺利发生该反应。 值得注意的是,当烷基硼酸作反应底物时,反应不能发生。 该反应的优点体现在底物作用范围广,操作简单经济,对环境友好,同时为构建含有C—Se键、C—Te键化合物提供了有效的方法。
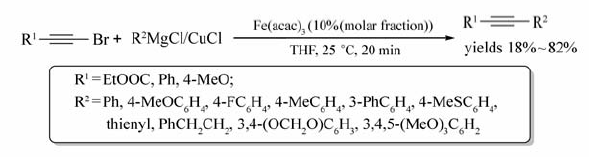
随着铁盐作为催化剂在有机合成中的应用逐渐增多,铁的另一种化合物Fe(acac)3也被广泛用于交叉偶联反应之中。 2010年,Daniele及同事[45]实现了1-溴炔与有机铜试剂的交叉偶联反应(Scheme 26)。反应以格氏试剂为原料来制取有机铜试剂,以THF为溶剂,Fe(acac)3为催化剂,25 ℃条件下作用20 min,以18%~82%的产率获取目标产物。使用有机铜试剂与格氏试剂相比,能有效减少底物自生偶联反应发生。该反应能合成一系列的芳基炔烃,具有反应条件温和,反应时间短等优点。值得注意的是1-溴乙炔与PhMgCl/CuCl在25℃作用时,得到的主要产物是烯烃(78%),在-20 ℃的条件下偶联炔烃的产率可达18%,相应的烯烃的产率则减少为56%。
Kumada偶联反应是指烷基或芳基格氏试剂与卤代芳烃或乙烯基卤化物在镍或钯催化下的交叉偶联反应[46]。 2014年,Francesca等[47]报道了Fe(acac)3催化嘌啉衍生物与格式试剂的Kumada反应(Scheme 27)。 该反应是合成帕金森病有效受体ST135中最为关键的一步,具有反应时间短(仅需1 h),条件温和,产物产率高(可达91%)的特点,该反应若用钯作催化剂则需反应16 h,产物的产率仅为85%。

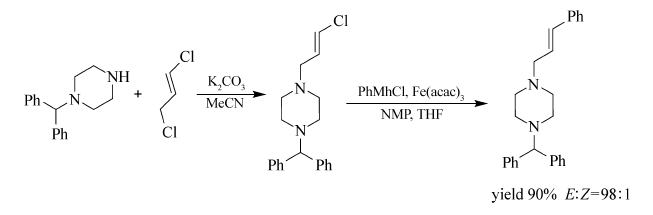
Shakhmaev等[48]通过1,3-二氯丙烯与哌嗪的 N-烷基化反应得到相应的烯基氯化物,并将其成功应用到Fe(acac)3催化下的交叉偶联反应之中(Scheme 28)。 通过与苯基氯化镁的偶联,其目标产物产率高达90%,且立体选择性较好( E:Z=98:2)。不过为保证目标产物的产率,需加入过量的苯基氯化镁。 该法具有反应条件温和,立体选择性高的特点。
以THF为溶剂,卤代烃在5%摩尔分数Fe(acac)3/TMEDA/HMTA(摩尔比1:2:1)组成的催化体系中,能够与烯基格氏试剂发生交叉偶联反应[49](Scheme 29)。 该反应底物适用范围广,不论单取代( α-或 β-取代)或二取代( α, β-或 β, β-二取代)烯基溴化镁,还是仲烷基溴化物(环状或无环状)均可发生该反应,并以48%~84%的产率获取产物。 相比碘代烃和溴代烃,氯代烃参与的反应所得的目标产物的产率较低。 该反应具有反应条件温和,底物适用范围广,所得产物立体选择性高的优点( Z:E=85:15~97:3)。

2010年,Matthias等[50]成功地实现了Fe(acac)3催化芳基格氏试剂的烯丙基化反应(Scheme 30)。 芳基溴化物在THF中与Mg/LiCl反应制取格氏试剂,进而与相应的烯丙基类化合物作用生成相应的目标产物。 研究表明,含有卤素、OTs、OP(O)(OEt)2、SMe、OAc等基团的烯丙基类化合物能与相应的格氏试剂发生反应;含酯基、卤素、胺基、嘧啶等的格氏试剂也能与烯丙基化合物作用。 该体系反应条件温和,操作简单,为苯丙烯类衍生物的合成提供了较为简便的方法。

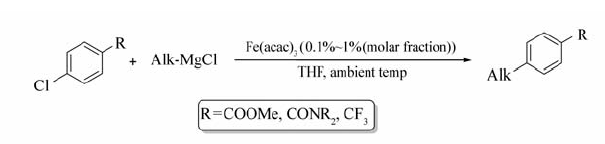
Philip等[51]报道了Fe(acac)3催化的氯代芳烃与烷基格氏试剂的偶联反应(Scheme 31)。 反应以TMEDA作为配体,在0 ℃条件下仅需较少的催化剂(摩尔分数0.1%)即可获取高产的目标产物。 含有CF3、酯基或酰胺基取代基的芳基氯化物均可顺利参与该反应之中。
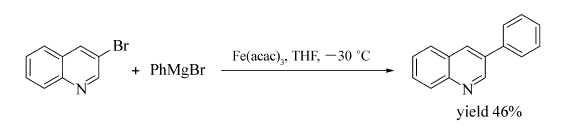
杂环卤化物与芳基格氏试剂的芳基化反应也可通过铁盐催化实现[52](Scheme 32)。 研究发现,不同溶剂对3-溴喹啉与苯基溴化镁反应的产率影响很大,THF/NMP为溶剂,反应在-30 ℃时以35%的产率形成相应产物;改用其他溶剂与THF组成共溶剂时,不仅偶联产物的产率有所降低,且还会得到3-溴喹啉的还原产物;若以THF为溶剂,反应不仅在-30、-20、-10 ℃和室温,甚至在-78 ℃均可发生反应,以32%~46%的产率得到目标产物。 最终选定以THF为溶剂,Fe(acac)3为催化剂,-30 ℃的反应温度为反应最优条件,该条件下2-氯喹啉、2-溴嘧啶、3-甲基-2-溴喹啉均能与苯基溴化镁反应。
Sushant等[53]报道了Fe(acac)3催化二卤代(杂环)芳烃与格氏试剂形成烷基(杂环)芳烃的反应(Scheme 33)。 体系中二卤代杂环芳烃烷基化的位置与卤素的位置、溶剂的性质以及格氏试剂的亲核性有关。 催化剂Fe(acac)3与其它金属化合物(钴,铜,镍)相比,不仅获取的产物产率高,而且选择性更好。

Jamie等[54]通过铁盐催化2-溴呋喃与格氏试剂的偶联反应,实现了2-取代呋喃的合成(Scheme 34)。 研究发现,使用Fe(bdm)3、Fe(dpm)3等铁盐作催化剂时,获得的2-取代呋喃的产率很低,使用FeF3·H2O作催化剂时,反应不能发生。 经过探究发现反应的最佳条件为:以Fe(acac)3作催化剂,1,3-二甲基-3,4,5,6-四氢-2-嘧啶酮(DMPU)为溶剂,反应温度为-25 ℃。 烷基格氏试剂(环状或非环状)、芳基格氏试剂均能与相应的底物有效地发生该反应。与钯催化剂相比,铁催化剂不仅经济环保,而且可实现伯烷基、仲烷基格氏试剂的反应。

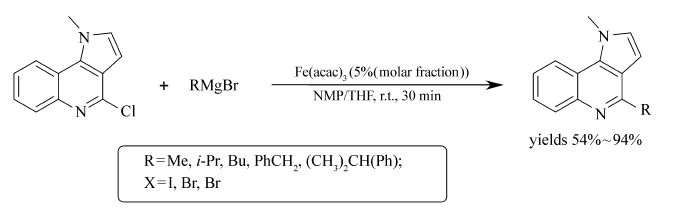
Evelina等[55]通过Fe(acac)3催化2-氯代喹啉衍生物与格氏试剂的反应,实现了2-烃基喹啉衍生物的合成(Scheme 35)。室温下作用30 min即可以54%~94%的收率获取相应的目标产物。 烷基或芳基格氏试剂均可顺利地完成反应,为构建 sp2- sp2和 sp2- sp3类型的碳碳键提供了便捷的方法,在药物化学具有广泛的应用前景。
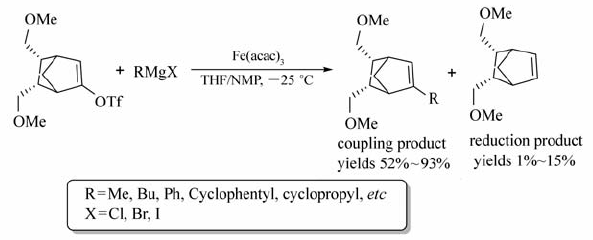
2008年Paul等[56]以Fe(acac)3为催化剂,在DMPU或THF/NMP中实现了双环烯基三氟甲磺酸酯与格氏试剂的交叉偶联反应(Scheme 36)。 在-25 ℃的条件下,以52%~93%的产率获取相应的2-取代环烯烃。 该法既可有效减少双环烯基三氟甲磺酸酯还原产物的形成,也能最大产率地获取目标产物。 同样条件下,Pd催化剂不能实现该转化。
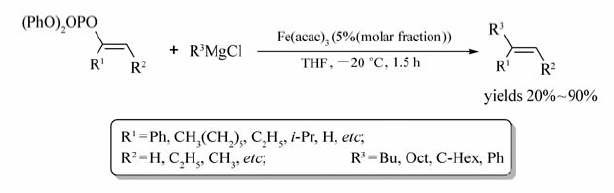
Gérard等[57]发现了Fe(acac)3能够成功地催化烯醇磷酸酯与格氏试剂的偶联反应(Scheme 37)。 反应使用铁盐作催化剂不仅经济环保,而且实现了反应的高度立体选择性。 值得一提的是,由醛或酮制备的烯醇磷酸酯在THF中即可发生该反应,而 α, β-二取代的烯醇磷酸酯则需加入助溶剂DMP或DMPU才能使反应有效地进行。 此外,格氏试剂的用量影响着产物的产率,如将BuMgBr由0.85化学计量增加到1.2化学计量,相应产物的产率可由56%( Z:E=91:9)提升至71%( Z:E=91:9)。 烯醇磷酸酯的烃基上含有氯、氰基、酯基等官能团时,反应能以84%~96%的产率获取产物,但当烃基上的官能团为溴原子时,体系得到的是烃基溴发生 β位消除的产物。
Knochel等[58]发现芳基酰氰化物在Fe(acac)3催化下能与芳基格氏试剂发生偶联(Scheme 38),以66%~98%的产率得到二芳基酮。 进一步研究表明杂环酰氰化物也可发生该反应,该法为二苯甲酮类化合物的合成提供了重要参考。

Chandra等[59]报道了Fe(acac)3催化烃基磺酰氯与格氏试剂的交叉偶联反应(Scheme 39)。 该反应以THF为溶剂,在室温无配体的条件下,作用3~6 h即可以较好的产率(56%~91%)获取相应目标产物。 各类芳香族、脂肪族或杂环格氏试剂均可参与到反应之中。 该反应的优点体现在无需任何配体,且条件温和,使用无害溶剂便可获取高产的目标产物。

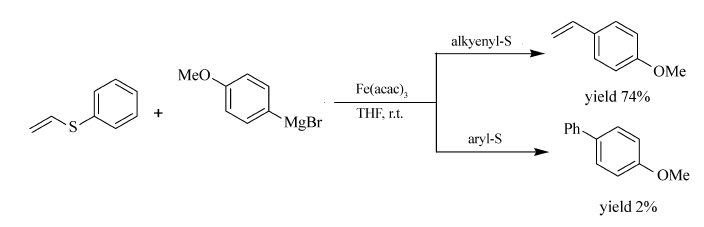
Itami等[60]成功地实现了Fe(acac)3催化的芳基硫醚与格氏试剂的交叉偶联反应(Scheme 40)。 该反应的最大特点体现在,反应在烯硫键处断裂与格氏试剂形成取代烯烃,而不是发生在芳基硫键上。
Fe(acac)3也可顺利催化芳基三甲基铵三氟甲磺酸盐与烷基格氏试剂的交叉偶联反应[61](Scheme 41)。 经过探究发现该反应的最佳条件为:Fe(acac)3作催化剂,THF为溶剂,NMP为共溶剂( V(THF): V(NMP)=7:1)。 值得注意的是共溶剂NMP的存在可提高该反应速率,对该反应至关重要。 含有 β氢的伯、仲烷基格氏试剂,以及含COOEt、COO i-Pr、COON-Et2、CN、CF3和嘧啶环的芳基三甲基铵三氟甲磺酸盐均可顺利参与该反应。
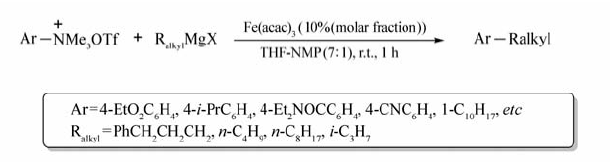 | Scheme 41 Iron-catalyzed cross-coupling of aryltrimethylammonium triflates and alkyl Grignard reagents[61] |
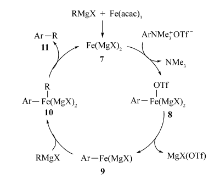
根据实验,Guo等[61]推测该反应的可能机理是Fe(acac)3先被格氏试剂还原形成活性催化剂Fe(MgX)2(化合物7),该活性催化剂与芳基三甲基铵三氟甲磺酸盐发生氧化加成形成中间体8与NMe3,接着化合物8分解产生活性中间体ArFe(MgX)(化合物9)和MgX(OTf),化合物9与RMgX形成中间体ArFe(R)(MgX)2(化合物10),该中间体经还原消除得到目标产物11和活性催化剂7(Scheme 42)。
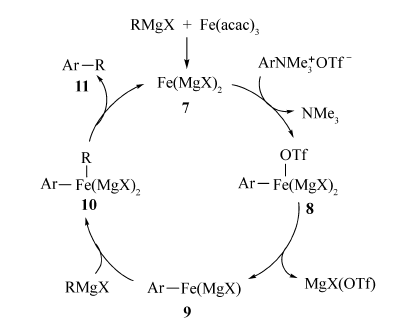
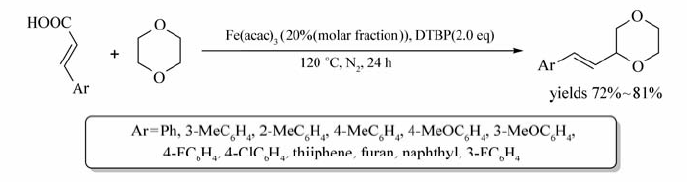
Zhao等[62]以二叔丁基过氧化物(DTBP)为氧化剂,实现了Fe(acac)3催化环醚的C—H键活化,反应在N2气氛围中,能以较好的产率获得目标产物(72%~81%)(Scheme 43)。 含有取代基的苯丙烯酸、杂环丙烯酸、1-萘基丙烯酸均可与1,4-二氧六环反应。此外[63],脂肪酸衍生物也可通过Fe(acac)3/Xantphos实现伯/仲烷基卤化物与格氏试剂的偶联反应。
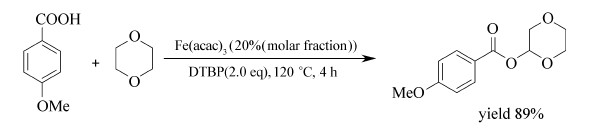
Fe(acac)3/DTBP体系也适用于羧酸与环醚的的交叉偶联脱氢反应,合成相应的酯类化合物[64]。 在120 ℃条件下,Fe(acac)3能催化氧化环醚的C( sp3)—H键发生氧化脱氢偶联反应生成相应的 α-酰氧基醚,目标产物最高产率可达98%(Scheme 44)。 对称或不对称的二元环醚、芳基羧酸、杂环羧酸均可顺利地发生该反应。 该反应具有底物适用范围广、反应产物产率高、区域选择性好的特点。
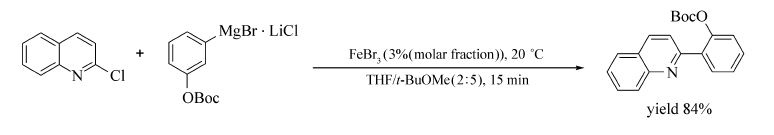
2012年,Olesya等[65]报道了FeBr3催化氮杂环氯化物或溴化物与芳基镁试剂的交叉偶联反应(Scheme 45)。 体系使用THF和 t-BuOMe的混合溶剂( V(THF): V( t-BuOMe)=2:5),这样既可避免底物的自身偶联副产物的形成,又可使底物最大限度地转化为相应的目标产物(产率60%~93%)。 该反应的底物适用范围广,含有二甲基胺基、甲氧基、叔丁氧羰基(OBoc)等官能团的芳基镁试剂均可发生该反应。
 | Scheme 45 FeBr3 catalyzed cross-coupling of N-heterocyclic chlorides with arylmagnesium reagents[65] |
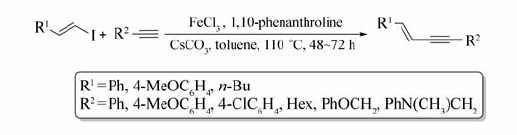
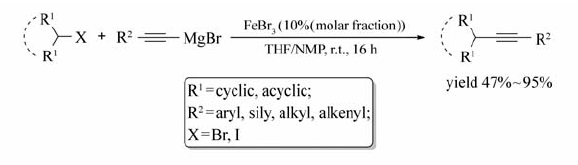
Chi等[66]实现了室温下FeBr3催化仲烷基卤代烃与炔基格氏试剂的交叉偶联反应(Scheme 46)。 无论环状或者非环状的仲烷基卤代烃、炔基溴化镁或含杂原子的炔基溴化镁均能发生该反应,目标产物的产率可达47%~95%。 值得一提的是,将溶剂改为THF/ O-TMEDA,催化剂FeBr3摩尔分数由10%增加至20%,可实现低活性的伯卤代烃与炔基溴化镁的偶联。 反应条件温和,并使用对环境友好、廉价的铁催化剂的优点使该反应在制药工程和材料化学工程具有潜在的应用前景。
除此之外,碳-杂键的构建也可通过FeBr3催化实现。 2014年,Cheng等[67]实现了无溶剂条件下亚磺酰亚胺与二芳基甲烷的交叉脱氢偶联反应(Scheme 47)。 反应以亚磺酰亚胺和二苯基甲烷为例对反应条件进行了探究,发现铜化合物、钯化合物或Fe(acac)3等催化剂对该反应无促进作用,而FeBr3的催化效果较好。 当反应底物的氢供体的取代基有一个苯环时,得到的产物为含1:1对映体的混合物。 该法避免了溶剂的使用,进一步向对环境友好,经济的绿色合成靠近。

Zhang等[68]通过苯环上的C—H活化,实现了均三甲氧基苯与二硫化物的氧化偶联反应(Scheme 48)。 研究发现,FeBr3为催化剂时,空气作氧化剂比K2S2O8、TBHP、O2等物质作氧化剂时的效果要好,并且更加经济、环保。 当二硫化物的苯环上含有甲氧基、硝基、氟、氯、溴等取代基时,反应能顺利的发生;杂环芳烃、1,3-二甲氧基苯、1,3,4-甲氧基苯也能与PhSSPh发生相应的反应。

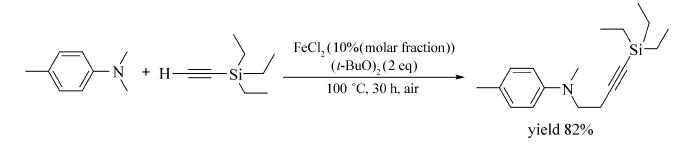
Chandra首次报道[69]了铁催化氧化叔胺与末端炔的氧化偶联反应(Scheme 49)。 Fe(acac)3、Fe(OAc)2、Fe(ClO4)2做催化剂时,该反应不能发生,Fe2(CO)9、Fe(CO)5做催化剂时,反应产率较低。 选用FeCl2作催化剂,( t-BuO)2作氧化剂,无需任何配体和溶剂,脂肪炔、芳基炔或三乙基乙炔硅烷均可发生该反应。 研究显示,当 N, N-二甲基苯胺的苯环有甲基、溴原子等取代基时,与脂肪炔烃或芳香炔烃反应均可以中等至较好的产率获取目标产物(47%~93%),当 N, N-二甲基苯胺做底物时,与芳香炔反应获取产物的产率较低(24%~32%)。
通过苯并咪唑与 N, N-二甲基乙酰胺的反应也能实现C—N键的构建[70](Scheme 50)。 以C6H5Cl为溶剂,FeCl2为催化剂,DTBP为氧化剂,在N2气氛围下,反应3 h,相应目标产率高达98%。 该反应的优点表现在: 1)底物范围广:各类的酰胺、磺酰胺均可做唑类化合物 N-烷基化的底物; 2)反应能从C—H键直接构建C—N键; 3)使用的催化剂经济环保。

Amanda等[71]实现了FeCl2催化芳基氨基磺酸酯或芳基氨基甲酸酯的烷基化反应(Scheme 51)。 反应以SIMes·HCl为配体,在THF/CH2Cl2体系中能以48%~99%的产率获取目标产物。 烷基氯化镁或杂环的氯化镁试剂均可实现该反应,该反应为构建 sp2 -sp2、 sp2 -sp3的碳碳键和合成含多种官能团的芳烃提供了有效的方法。

Peng等[72]首次实现了在存在 N-杂环配体时,铁催化低活性的富电子芳基氯与烷基格氏试剂的反应(Scheme 52)。 通过对铁化合物的探索发现FeCl2(H2O)4比FeCl2更适合作该体系的催化剂。 伯烷基格氏试剂能获取较高产率的目标产物,而环状的仲烷基格氏试剂获取的目标产率较低,并且无环的仲烷基格氏试剂在获取目标产物的同时还易形成副产物正烷基异构体。 副产物的形成表明对该反应而言还原消除与 β消除在同时进行。

Krishna等[73]在2007年成功地实现了Fe(OAc)2催化的卤代烃与格氏试剂的 sp3 -sp3交叉偶联反应(Scheme 53)。 反应若不加入配体,或铁盐选择不当,会产生较多的副反应(反应物的自偶联、歧化、 β-消除反应等),通过对反应条件的筛选,最终选定4,5-双二苯基膦-9,9-二甲基氧杂蒽(Xantphos)作配体,Fe(OAc)2作催化剂,乙醚为溶剂,以43%~64%的产率得到相应的目标产物。

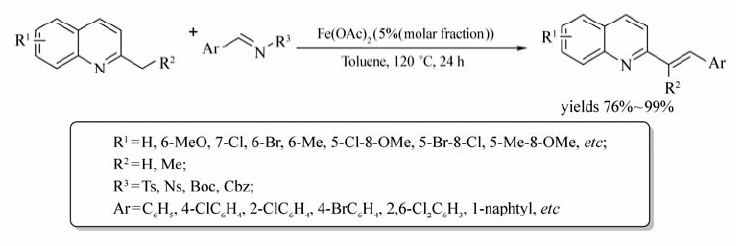
Qian等[74]首次报道了Fe(OAc)2催化 N-杂环芳烃与 N-磺酰亚胺的交叉偶联反应(Scheme 54)。 该反应实现 N-杂环芳烃中的
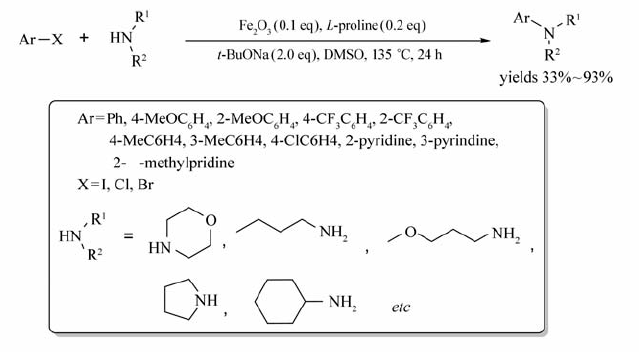
通过改变铁盐和配体的种类,其它 N-烷基化反应也可顺利进行。 以DMSO为溶剂, L-脯氨酸为配体,Fe2O3为催化剂,可实现卤代芳烃与胺的 N-芳基化反应[75](Scheme 55)。 该反应无需隔绝空气和水,底物适用范围较广,芳杂环卤化物也可参与该反应,产物产率高达93%。 以2-三氟甲基碘苯与对硝基苯胺做反应底物时,得到的产物产率较低。 虽然 L-脯氨酸和Fe2O3对该反应能否顺利进行至关重要,但是少数底物在不存在催化剂和配体时也可发生反应(如环丁胺与对氯碘苯),只不过获取的产物产率均明显偏低。
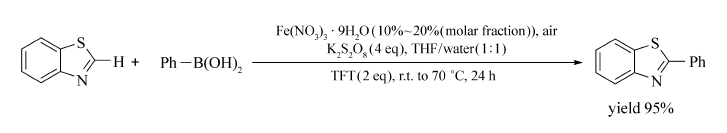
以Fe(NO3)3·9H2O/K2S2O8为催化体系,THF/H2O(体积比1:1)为溶剂可实现杂环化合物、醌与芳基硼酸的偶联反应[76](Scheme 56)。 室温至70 ℃时含有一个杂原子或两个杂原子的化合物(如喹啉、喹喔啉、苯并噻唑等)均可发生芳基化反应。 获取产物的产物分别为43%~92%、54%~95%。 在室温条件下,醌也能与芳基硼酸也能以较高的产率得到相应目标产物(60%~90%)。
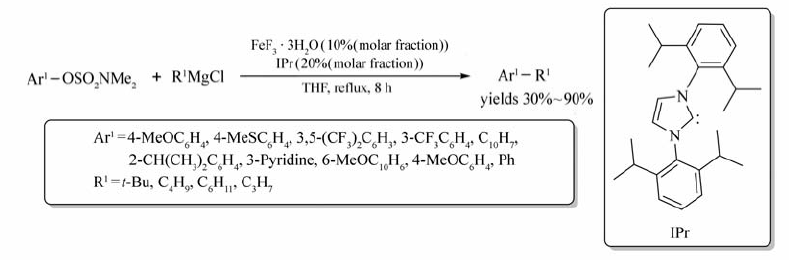
以THF为溶剂,FeF3·3H2O可催化芳基胺基磺酸酯与烷基格氏试剂的交叉偶联反应,以39%~90%的产率获取相应的目标产物(Scheme 57)[77],该条件下也能实现甲基磺酸酯与烷基格氏试剂的反应。 与其它催化剂相比,该反应使用FeF3·3H2O作催化剂,既能避免异构产物的形成,同时还能最大限度地获取相应的目标产物。 2014年Anrawal等[78]首次将芳基格氏试剂用作上述反应的底物,并且可获取高产率的目标产物。 相比烷基氯化镁,芳基溴化镁的使用能有效地避免格氏试剂的自身偶联反应的发生。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
由于易于获取、环境友好、无毒、廉价的特点使得铁催化剂在有机合成中备受关注,本文主要总结了铁催化剂在构建C—C、C-杂原子等偶联反应中的应用。 然而当交叉偶联反应以铁为催化剂时存在下面的问题:1)大部分铁盐对空气湿气敏感,反应要求在无水无氧条件下进行;2)在催化硫醇、酰胺、胺与炔烃的偶联反应时,催化剂的纯度以及来源对反应产物均会产生较大的影响[79]。 虽然存在这样的问题,但总的来说,铁催化的偶联反应反应还是取得了巨大的成功:化学选择性高,产物的产率较好,底物的官能团耐受性好,避免了在反应过程中保护官能团,若将其成功实现工业化,必将会带来较大的经济效益。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|
| [74] |
|
| [75] |
|
| [76] |
|
| [77] |
|
| [78] |
|
| [79] |
|