
DING Fanshu, NIE Xiaowa, LIU Min, et al. Research Progress in Catalytic Conversion of Carbon Dioxide to C2+ Hydrocarbons over Fe-Based Catalysts [J]. Chinese Journal of Applied Chemistry, 33(2): 123-132


将二氧化碳(CO2)催化加氢转化为具有高附加值的烃类化合物,既可减缓大气中CO2浓度的攀升速度,又符合可持续发展战略,对环境和社会均具有重要意义。本文综述了Fe基催化剂上CO2加氢制C2+烃的研究进展,着重介绍了反应路径及机理、催化剂研制及反应器设计,展望了CO2制烃的研究前景。
Catalytic conversion of carbon dioxide(CO2) to value-added hydrocarbons is of great environmental and social importance, which can not only reduce CO2 concentration in the atmosphere, but also conform with sustainable development strategy. This paper reviews the progress in catalytic conversion of CO2 to C2+ hydrocarbons over Fe-based catalyst. Reaction pathway and mechanism, catalyst preparation and reactor design are emphatically introduced. In addition, the future of hydrocarbons synthesis via CO2 hydrogenation is also summarized.

CO2是大气中一种无色无味的气体,是碳基化合物完全氧化的最终产物。在大自然中,植物消耗CO2并在太阳光的帮助下进行光合作用,人类这一有机生命体在消耗有机食物的同时,通过呼吸作用排放CO2。以煤、石油和天然气为代表的化石燃料的大量消耗推动了社会的迅猛发展,也导致了CO2排放量日益攀升,与工业革命前的280×10-6相比,2012年大气中CO2浓度达到394×10-6,比18世纪中期高出40%[1]。另一方面,化石燃料的逐渐耗尽,导致了石油价格的急剧波动。将CO2催化加氢合成高附加值的烃类化合物,既提供了一种CO2综合利用的有效途径,又推动了新型洁净能源技术的发展,具有极大的研究前景,近年来引起了国内外学术界的广泛关注[2]。
Fe催化CO2加氢制烃反应与传统CO加氢的费托合成工艺(Fischer-Tropsch synthesis, FTS)具有诸多相似性,因此,一部分研究者将其称为CO2费托合成[3,4]。1925年,由CO加氢合成烃类化合物作为低硫柴油原料的FTS反应,提供了克服基于石油的烃类供应或成本问题的新技术。1957年,Hall等[5]通过对FTS反应的副产物CO2进行同位素标定,基本确定Fe基催化剂上由CO2一步加氢合成烃类这一反应途径并不可行。1978年,Dwyer等[6]发现FTS反应过程中的副产物CO2会对Fe基催化剂的活性和产物分布产生影响,这是Fe基催化剂上CO2加氢反应的最早研究报道。Riedel等[7]通过改变停留时间,对100Fe/13.3Al2O3/10.7Cu/8.9K催化剂上CO2加氢制烃反应进行动力学分析,认为CO2一步加氢制烃反应在理论上可以发生,但这一结论尚不能成为单一活性位上CO2直接加氢制烃的本征反应机理的证据。其它金属如Co、Ni、Ru和Pd等,也被应用于CO2制烃反应中,但其主产物为CH4,只有很少量的C2+烃生成[8,9,10]。
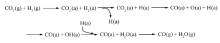
随着研究的深入,在Fe基催化剂上以CO为中间产物,如式(1)、(2)所示的两步反应串联机理,逐渐被研究者认可:第1步为逆水煤气变换(Reverse Water Gas Shift,RWGS)反应,CO2加氢转化为CO,催化效率受反应平衡限制和动力学控制;第2步为FTS反应,将第1步反应生成的CO进一步加氢生成烃类化合物,反应受动力学控制[11,12,13,14]:
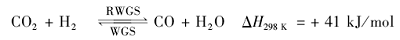 |
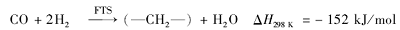 |
Willauer等[15]对固定床上Fe催化CO2制烃反应进行了模型构建和动力学模拟,结果表明,FTS反应速率远低于RWGS反应速率,FTS反应是CO2制烯烃总反应的速率控制步骤。如果能够通过RWGS反应将CO2完全转化为CO,则CO2加氢反应产物可符合FTS反应研究中常用的Anderson-Schulz-Flory(ASF)分布,烃类产物选择性极大值分别可达到汽油馏分(C5~C11)45%,柴油(C12~C20)30%,低碳烯烃(
明确Fe基催化剂上RWGS和FTS反应机理对CO2加氢制烃反应的催化剂设计及反应条件的确定具有重要意义。RWGS的反应机理主要有氧化还原机理和中间产物分解机理[17]。He等[18]根据DFT计算结果认为,在Fe3O4(111)表面,氧化还原机理是主要的反应机理(如Scheme 1所示),CO2在Fe3O4(111)表面被还原为CO,中间产物甲酸盐HCOO和羧酸基COOH分解机理较难发生,八面体Fe原子在CO2分子的吸附和活化过程中起着重要作用。在后续反应中CO进一步FTS加氢生成有机烃。
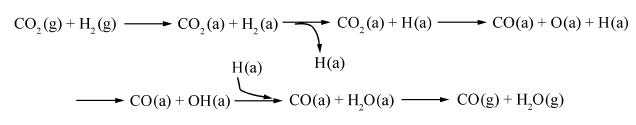 | Scheme 1 Reaction pathways of RWGS on Fe3O4(111)[18] |
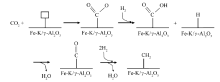
Lee等[19]认为, Fe基催化剂上CO2加氢反应过程中,催化剂表面CO2分子被Fe2+还原,形成Fe3+-COO·结构,H2分子解离为H自由基,当H自由基进攻羰基碳时,生成HCOOH和CO等副产物,Fe-CH2自由基为碳链增长的活性物种;在实际反应中,Fe催化CO2加氢主要产物是C2+烃,因此碳链增长被认为是主要反应途径。在Al2O3负载FeK催化剂上,这一反应途径同样适用(如Scheme 2所示)[19,20]。
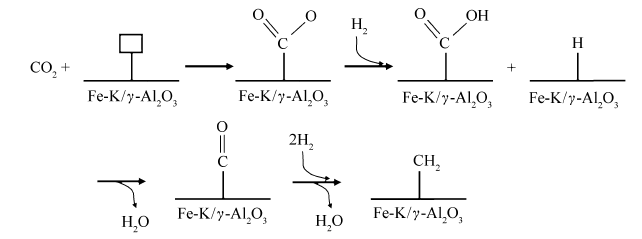
CO2制烃反应中,烃类产物主要由第2步FTS反应中碳链增长生成。Ziegler等[21]用第一原理方法计算了FTS中Fe(100)晶面上碳链增长过程,假定丙烯和丙烷为目的产物,发现最稳定的C3表面物种为CCCH2和CCCH3,而Fe(110)上的碳链增长反应是由C和CCCH2/CCCH3的复合引起的,而非表面的乙烯基和亚甲基耦合反应所致。Cheng等[22]用DFT方法计算了Ru、Fe、Co、Rh和Re表面在FTS碳链增长过程中C-C耦合过渡态和能垒,结果表明,在不同金属表面均具有相似的过渡态结构,但能垒相差很大;Rh和Ru表面的碳链增长主要是通过C+CH和CH+CH进行,Co表面为CH2+CH2和C+CH3,Fe表面为C+CH3,而Re表面为C+CH实现C-C耦合。
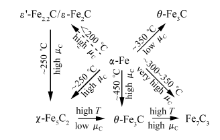
由CO2制烃的反应途径和碳链增长机理可知,FTS反应的活性物种对CO2制烃反应中长碳链烃的生成具有重要意义。1980年,Nlemantsverdrlet等[23]通过穆斯堡尔谱和X射线衍射技术证实,一种常被归属为六方晶系的Fe2C的碳化铁物种实际为 ε`-Fe2.2C,在反应过程中生成的未知碳化铁物种Fe xC是介于 α-Fe和晶体碳的一种低度有序结构,其相对含量与催化剂活性有对应关系。Riedel等[24]在反应压力1 MPa、反应温度250 ℃、H2/CO2=3条件下,研究了Fe-Al-Cu-9K催化剂上氧化铁随反应时间的相态组成变化,发现在反应初期,主要发生反应物在催化剂表面的吸附和碳化;随着反应时间逐渐延长,主要发生RWGS反应,积碳开始生成;进一步延长反应时间,开始发生FTS反应并逐渐达到稳态;Fe2O3、Fe3O4以及 α-Fe的相对含量均随着反应时间的延长而降低,碳化铁物种Fe5C2含量逐渐增加。对照以上结果Riedel推测 α-Fe不是FTS的活性中心,而Fe5C2很有可能是FTS生成高碳烃的活性物种。Bae等[25]对共沉淀制备的K/FeCuAlO x催化剂上FTS反应进行了系统研究,认为Fe物种的相态变化为 α-Fe2O3→Fe3O4→ α-Fe→ χ-Fe5C2,Fe基催化剂上铁的碳化物种为反应活性位。Ding等[26]利用穆斯堡尔谱分析了Fe基催化剂在FTS过程中含碳物种的相互转化,碳化过程中 α-Fe2O3首先被还原为Fe3O4,继而在催化剂表面碳化为原子形态(C α)和聚合物形态(C β)两种含碳物种;随着反应的进行,近表面区域的Fe3O4逐渐转化为 χ-Fe5C2,此时C α和C β含量呈上升趋势;一部分C α和C β两种含碳物种也会合并为石墨化的碳化物—C δ,C δ物种会大面积覆盖催化剂表面,导致催化剂活性的降低,而 χ-Fe5C2的生成以及催化剂表面的C α、C β物种改善了催化剂活性。Scheme 3为Emiel等[27]利用从头计算方法得到的碳化铁结构及其转化途径,可以看出,CO加氢反应过程中铁的碳化物存在 ε-Fe2C、 ε`-Fe2.2C、Fe7C3、 χ-Fe5C2和 θ-Fe3C等几种形态,并在一定条件下可以相互转化。Mogorosi等[28]则认为,在FTS反应中, α-Fe主要转变为 χ-Fe5C2,而FeO转变为非活性的 ε-Fe2C。Ma等[29]以Fe(CO)5为铁源,通过引入溴离子提高铁的抗氧化能力,成功合成了Fe5C2纳米粒子并将其应用于FTS中;与传统的 α-Fe2O3为Fe源的催化剂相比,Fe5C2在反应初期就具有很高的反应活性,而经过预还原的 α-Fe2O3催化剂,需要经过一段时间的活化,其活性才能达到稳定。
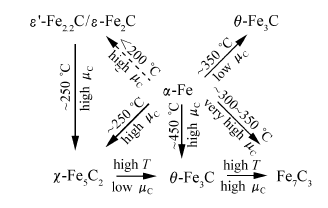 | Scheme 3 Qualitative interpretation of ab initio atomistic thermodynamics study of the iron carbide structure[27] |
活性金属的存在形式和还原能力与催化剂的制备方法密切相关。Huang等[30]利用穆斯堡尔谱对不同方法制备的Fe基催化剂进行了系统研究,结果表明,熔融法和沉淀法制备的Fe催化剂均是一种晶粒较大的本体Fe催化剂,前者主要Fe相为Fe3O4,部分为FeO,后者为 α-Fe2O3,两个催化剂均具有较好还原性能,几乎可完全还原为 α-Fe; γ-Al2O3负载的Fe基催化剂是一种高分散的催化剂,由两个近似等量的Fe相组成,一相是部分被Al离子替代的 α-(Fe1- xAl x)2O3,另一相则为Fe与载体Al2O3较强相互作用形成的Fe、Al、O化合物中的Fe3+相,负载型催化剂还原能力较差,且还原过程中形成新FeAl2O4相。Hou等[31]对共沉淀法制备的Fe/Cu/K/SiO2催化剂中K加入顺序进行了系统考察,结果表明,K助剂添加顺序对催化剂的比表面积、孔结构、孔径分布及晶粒大小影响很小,然而后加K的催化剂可使较多的K富集于催化剂表面,导致表面的有效K含量增多,抑制了H2的吸附,催化剂还原能力降低。Liu等[32]比较了共沉淀法、溶胶凝胶法和改进溶胶凝胶法制备的FeMo/SiO2催化剂的性质及FTS反应活性,改进溶胶凝胶法制备的催化剂具有更高密度的反应活性位,Fe的晶粒尺度更小,Fe-SiO2相互作用更弱,Fe物种更易被还原,催化剂具有更高的反应活性和更低的甲烷选择性,稳定性也得到了改善。Prasad等[33]认为浸渍法制备的Fe基催化剂在FTS反应中的失活主要是由于积碳引起的,而共沉淀法制备的催化剂的失活是由于反应过程中Fe物种晶粒尺寸的增长。
已有研究表明,结构性助剂的加入可以有效延缓催化剂的失活,增强催化剂耐磨性,提高催化剂的稳定性,并有可能通过金属-结构助剂相互作用进一步调变催化剂性能[34,35]。Hou等[36]用共沉淀法制备了100Fe/5Cu/4K/25SiO2催化剂,比较了Si(P)(共沉淀过程中加入SiO2)和Si(B)(作为粘结剂使用的SiO2)比例对催化剂性质及活性的影响,随着Si(P)/Si(B)比值的增加(0/25~15/10),催化剂的晶粒尺寸降低,表面酸性明显增加,催化剂的还原和碳化能力得到增强,催化剂活性升高;当增加Si(P)/Si(B)比值至25/0,金属-SiO2相互作用过强导致催化剂的还原和碳化行为受到抑制。Wan等[37]发现在沉淀过程中加入Al2O3,可以改善Fe物种的分散程度,加强Fe-Cu和Fe-K之间相互作用,催化剂的碳化能力和FTS活性都有所提高;与此相比,Al2O3作为粘结剂加入,增加了Fe-Al2O3相互作用,降低了催化剂的表面酸性,抑制了CO的吸附。机械混合法也是常用的催化剂制备方法之一,然而Yan等[38]发现在共沉淀法制备的Fe-Cu-Al-K催化剂中,机械混合Al2O3或SiO2均无法对催化剂活性有任何改善,并且机械混合SiO2会降低烯烃和长碳链烃类的选择性;在共沉淀过程中加入Al2O3,CO2转化率和C2+选择性都有所提高,CO选择性降低。
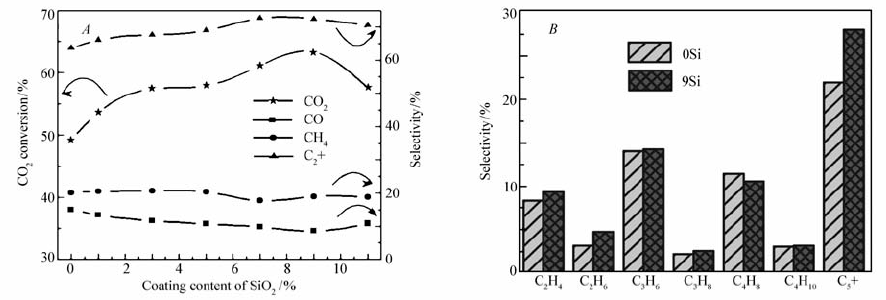
表面修饰法可以改变催化剂活性及产物分布。CO2制烃的两步反应均有副产物水产生,水蒸气在催化剂活性位上与反应物和反应中间体产生竞争吸附,影响催化剂活性。Ding等[39,40]对传统Fe-K/Al2O3催化剂进行疏水SiO2包覆处理,减弱了副产物水的竞争吸附,并且SiO2与活性金属、载体的相互作用加强了反应中间体CO的化学吸附,大大提高了催化剂活性和C2+烃选择性,CO2转化率和产物选择性变化趋势如图1所示,最佳SiO2包覆量为9%。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
碱金属助剂在CO2制烃反应中应用广泛,Kou等[41]以Fe/ZrO2为催化剂,得到68%的C2~C5烃选择性,但CO2转化率仅为20%;Wang等[42]对Fe/ZrO2进行碱金属改性,明显提高了CO2转化率,且产物中CH4和低碳烷烃选择性降低,当使用K+改性时,CO2转化率由32%升至43%,低碳烯烃占总烃产物比例由0.1%升至44%。Dry等[43]发现K助剂不仅可以通过增加催化剂的碱性以改善反应物CO2的化学吸附,提高催化剂活性,而且可以通过向Fe的缺电子d轨道贡献电子,加强中间产物CO在催化剂上的解离吸附,抑制氢的解离吸附,产物中烯烃选择性升高。Xu等[44,45]在K改性的Fe/Silicalite-2催化剂上得到了相似的转化率、选择性变化趋势。Nam等[46]发现,K助剂还可以改善催化剂的碳化能力,进而使C2-C4烯烃选择性由9%升高至32.4%,C5+选择性由1.5%升至44.5%,CO-TPR的表征结果证明,K助剂的加入,使催化剂的还原/碳化峰向低温方向移动,并且峰强度明显增加。Huo等[47]的理论计算结果表明,助剂K2O对Fe基催化剂上高度有序、高活性晶面如Fe(211)和Fe(310)晶面具有稳定化效应。Ding等[48]比较了K前驱体对FeK/Al2O3催化剂在CO2加氢制烃反应中的影响,发现KNO3和K2SiO3为较好的K前驱体,随着K负载量增加,CO选择性降低,C2~C4烯烃和C5+选择性增加,最佳负载量为10%。
在FTS和CO2加氢反应中,Mn助剂既是结构助剂,又可作为电子助剂改变Fe的电子密度,从而改善催化剂的还原能力、分散能力以及碳化能力[49]。Xu等[50]发现,Mn的加入可以改变反应产物分布,甲烷的生成受到抑制,产物的烯烷比由1.09增加至5.78;过量Mn的加入,会导致活性相的团聚,催化剂活性降低,非目的产物的选择性增加。Li等[51]认为,Fe-Mn-K/SiO2催化剂中的Mn助剂可以抑制 α-Fe2O3颗粒的增长,阻碍FeO在氢气气氛中进一步还原为Fe单质,适当的Mn加入可以增加催化剂表面酸性,促进催化剂的碳化。Dossary等[52]利用溶胶凝胶法制备了一系列不同Mn含量的 xMnFe( x=0,0.05,0.1,0.2,0.3,0.5)金属氧化物,其中0.05MnFe样品具有介孔结构,较大的比表面积,较低的还原温度和较高的C2+烃选择性。
Cu助剂和Mn助剂具有一定相似性,均可以改善催化剂的加氢能力,因而在Fe催化CO2加氢反应中,可以替代Mn作为助剂使用。Cu也是RWGS反应的催化剂之一,在催化剂碳化过程中,Cu可以控制Fe2O3的还原程度,增加其在催化剂表面的分散度[53]。King等[54]认为当CuO被还原为单质形态,可以提供解离氢吸附的活性位。Zheng等[55,56]使用尿素沉淀凝胶、机械混合和等体积浸渍法相结合的方法,制备了一系列的纳米尺寸FeK-M/Al2O3(M=Cd,Cu)催化剂,随着K含量由0%增加至10%,低碳烯烃选择性由3.1%增加至29.5%,Cd和Cu助剂可促进Fe物种的还原,改善目的产物的分布,其中3.8%Cu的加入使低碳产物烯烷比由3.64增加至5,加入1.1%Cd使C5+产物选择性由25%增加至28.5%。
在负载型催化剂中,载体可以使负载金属更好的分散,抑制其烧结团聚,同时其表面的酸碱性及孔道结构对催化剂的活性及产物选择性都有一定影响。氧化物载体在CO2加氢制烃反应中使用广泛,其中 γ-Al2O3为载体的催化剂活性最好,其次是SiO2和TiO2[38,57,58]。以Al2O3为载体的催化剂,较强的金属-载体相互作用有利于铁的分散,对金属的烧结具有抑制作用[59],而较小的金属-载体相互作用往往导致铁颗粒尺寸的增大[60]。Dorner等[61]认为,Al2O3可以和K助剂形成KAlH4活性相,而KAlH4在250~300 ℃对氢分子具有可逆吸附能力。Al2O3载体的物化性质对催化剂活性和产物分布也会产生很大影响,Ding等[62]以6种拟薄水铝石为原料制备了Al2O3负载Fe基催化剂,应用于CO2制烃反应,结果表明,Al2O3的等电点明显影响了负载金属的分散度和颗粒大小,随着等电点的增加,Fe分散度增加,CO2转化率和C5+烃选择性升高。Wang等[42]认为SiO2、Al2O3、TiO2、ZrO2、介孔碳和碳纳米管中,ZrO2为载体的催化剂具有最高的低碳烯烃选择性,碱金属离子改性(Na+、K+、Cs+)的Fe/ZrO2催化剂,可以明显提高CO2转化率、低碳烯烃和C5+烃选择性。
与传统的氧化物载体相比,分子筛载体由于具有规则的孔道和丰富的酸中心,在CO2和CO加氢转化反应中表现出独特的催化性能。Bai等[63]考察了Fe-Zn-Zr/分子筛复合催化剂上分子筛类型对CO2加氢反应性能的影响,结果表明,不同分子筛对复合催化剂性能的影响不同,与NaY、HZSM-5和HM相比,HY是合成异构烷烃有效的复合催化剂;分子筛的酸性及酸强度对复合催化剂性能有较大影响,中等强度和较高强度的酸性位有利于异构烃的生成。Nam等[46]以HY作为Fe基催化剂的载体应用于CO2加氢反应时,铁负载量为17%时,载体表面形成单层活性相分布,对HY载体进行碱金属离子交换可以提高催化剂表面碱性,CO2加氢活性提高。Hu等[64]将多孔锰钾矿型八面体分子筛K-OMS-2用于CO2制烃反应,比较表面负载Fe(SCI)和骨架掺杂Fe(FDI)两种催化剂反应结果,发现SCI具有很高的低碳烯烃选择性,但CO2转化率低于FDI。
有序介孔碳具有比表面积大、孔径分布均一、化学稳定性好等特点,在吸附、分离和催化等领域具有广阔的应用前景。Li等[65]改变热处理温度脱除模板剂,合成了具有不同表面性质的体心立方结构介孔碳材料,并以其为载体合成Fe/OMC催化剂用于CO2制烃反应,实验结果表明,高温(>600 ℃)脱除模板剂的Fe/OMC催化剂比低温(400 ℃)处理的样品具有更高的CO2转化率和C2+烃选择性,但产物中几乎没有烯烃生成,而OMC-400为载体时,C2~C4烯烃选择性达16%。Mu等[66]采用溶剂挥发诱导自组装方法制备了系列Fe基催化剂Fe-C- m,随着乙酰丙酮加入量的增加,Fe-C- m氧化铁颗粒减小,分散程度提高,在CO2加氢制烃反应中,Fe-C-0.27催化剂活性最好。Chew等[13]比较了含氧官能团碳纳米管(OCNT)、含氮官能团碳纳米管(NCNT)和SiO2负载Fe应用于CO2制烃反应的催化剂活性,其中Fe/NCNT还原温度最低,说明含氮官能团能够改善Fe的还原能力,Fe/SiO2上金属-载体相互作用最强,Fe还原温度最高;Fe/NCNT催化剂上C2~
文献报道的CO2加氢合成烃类反应使用的反应器绝大部分都是固定床反应器。Fujimoto等[67]采用RWGS-FTS两段式反应器工艺用于CO2制烃反应,在第二段反应器,脱除进料中第一段反应生成的副产物水,烃类产物的收率明显改善。实际应用中,流化床和浆态床反应器可以快速移走放热反应过程中生成的热量,传热传质速度快,反应相可以有效接触,从而烃类产物的生成率更高。虽然CO2加氢的第一步RWGS反应为吸热反应,但第2步FTS为强放热反应,因而使用流化床或浆态床反应器替代固定床反应器是合适的选择。Lee等[68]考察了流化床和浆态床上,Fe-K/Al2O3和Fe-Cu-Al-K催化剂在CO2加氢合成烃类的反应活性,研究表明,流化床和浆态床上催化剂活性高于固定床反应器。通过反应器参数的设定,可以选择性生成低碳链烯烃或者长碳链烃类化合物。浸渍法制备的K/FeO x催化剂抗磨损能力好,适合在鼓泡流化床反应器上使用[69]。Yan等[38]比较了悬浮床反应器和固定床反应器在CO2加氢反应中的活性差异,固定床反应器可以得到较高的CO2转化率和更低的CO选择性,同时C5+烃类选择性和烯烷比更高;悬浮床反应器中,反应物和在液态介质中传质的限制是导致CO2转化率和C5+烃类产物选择性较低的主要原因。
虽然CO2加氢制烃技术对改善环境和提供新型洁净能源都具有促进意义,但反应转化率和长碳链烃类选择性偏低、催化剂易失活、反应机理不明确等因素仍限制了这一技术的发展。未来的发展中,以下几方面研究尤为重要:1)加速发展电解水和太阳能光解水技术,使生产大量而廉价的氢成为可能,进而解决CO2加氢过程中氢源供给问题;2)设计开发新型催化剂,如将少量Co引入传统Fe基催化剂中制备双金属催化剂[70,71,72],或将活性金属封装进入中空分子筛中,利用分子筛孔道的限域作用,提高目的烃类的选择性;3)从反应器设计入手,效仿Farsi等[73]的工作,将水蒸气选择透过性膜反应器应用于Fe基催化剂上CO2加氢制烃的研究中,及时移走反应过程中生成的副产物水,提高CO2转化率;4)加强反应机理的研究,利用原位表征技术,检测催化反应过程中催化剂组成、结构变化和反应物吸附形式,进一步辅助催化剂开发和反应器设计。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|
| [58] |
|
| [59] |
|
| [60] |
|
| [61] |
|
| [62] |
|
| [63] |
|
| [64] |
|
| [65] |
|
| [66] |
|
| [67] |
|
| [68] |
|
| [69] |
|
| [70] |
|
| [71] |
|
| [72] |
|
| [73] |
|