
REN Lilei, PENG Xiaoxia, ZHAO Xiuli, et al. Synthesis and Antitumor Activity of Porphyrin Modified with 5-Fluorouracil[J]. Chinese Journal of Applied Chemistry, 33(12): 1415-1419


合成了一种5-氟尿嘧啶修饰的自由卟啉(5-[2-(5-氟尿嘧啶-3-基)乙氧基苯基]-10,15,20-三(4-甲氧基苯基)卟啉)及其2种金属卟啉配合物:5-[2-(5-氟尿嘧啶-3-基)乙氧基苯基]-10,15,20-三(4-甲氧基苯基)锰卟啉和5-[2-(5-氟尿嘧啶-3-基)乙氧基苯基]-10,15,20-三(4-甲氧基苯基)锌卟啉。 通过紫外可见光谱(UV-Vis)、红外光谱(IR)和核磁共振谱氢谱(1H NMR)对目标化合物进行了结构表征。 用噻唑蓝法(MTT法)测定了自由卟啉、锰卟啉及锌卟啉分别对肺腺癌细胞株A549、肝癌细胞株Bel7402和人结肠癌细胞株HCT-8的抑制活性。 其中,锰卟啉对人结肠癌细胞株HCT-8的半抑制浓度为IC50为17.8 mg/L,具有一定的细胞毒作用。
The porphyrin modified with 5-fluorouracil(5-[2-(5-fluorouracil-3-yl)-ethoxyphenyl]-10,15,20-tri(4-methoxyphenyl) porphyrin) and its two metal porphyrin complexes: 5-[2-(5-fluorouracil-3-yl)-ethoxyphenyl]-10,15,20-tri(4-methoxyphenyl) Mn porphyrin and 5-[2-(5-fluorouracil-3-yl)-ethoxyphenyl]-10,15,20-tri(4-methoxyphenyl) Zn porphyrin were synthesized. The structures of the three compounds were characterized by ultraviolet visible spectroscopy(UV-Vis), infred spectroscopy(IR) and proton nuclear magnetic resonance spectroscopy(1H NMR). The antitumor activities of these compounds to A549, Bel7402, HCT-8 were tested using the methyl thiazolyl tetrazolium(MTT) method. The results show that the IC50 value of Mn porphyrin to HCT-8 is 17.8 mg/L.
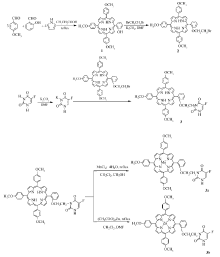
5-氟尿嘧啶(5-FU)是临床应用的广谱抗肿瘤药物,其主要缺点是服药有效剂量与中毒剂量相近,在杀死癌细胞的同时正常细胞损伤也较严重。 为此,科研工作者对5-FU进行了大量的化学修饰并取得显著效果[1,2,3]。 利用卟啉对癌细胞有特殊的亲合性,能够在癌细胞中有选择性地滞留[4,5],本文将卟啉与5-FU连接起来,合成了一种卟啉-5-氟尿嘧啶(3)及金属锰卟啉(3a)和锌卟啉(3b),并通过紫外可见光谱(UV-Vis)、红外光谱(IR)和核磁共振谱氢谱(1H NMR)确证了产物的结构。 初步测试了它们对肺腺癌细胞株A549、肝癌细胞株Bel7402和人结肠癌细胞株HCT-8的抑制活性,得出了一些有意义的结论。
合成路线如Scheme 1所示。
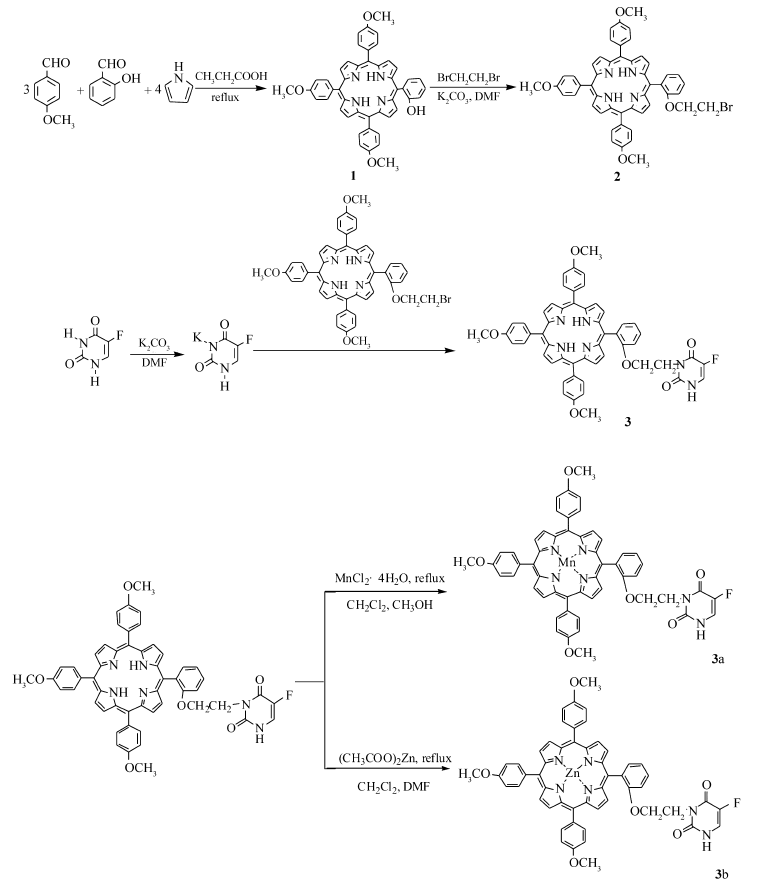 | Scheme 1 The synthetic route of the inter mediates and target compounds |
{kind=link}
UV-2501(PC)S型紫外-可见分光光度计(日本岛津公司);FTIR8900型傅里叶变换红外光谱仪(日本岛津公司);Mercury-400型核磁共振仪(美国Varian公司)。
5-氟尿嘧啶,生化试剂,购自百灵威试剂公司,其它试剂均为分析纯,购自天津北方天医化学试剂公司;吡咯和1,2-二溴乙烷使用前重蒸;5-氟尿嘧啶,用水重结晶,mp 281 ℃;碳酸钾使用前焙干;5-邻羟基苯基-10,15,20-三(对甲氧基苯基)卟啉(1)和5-邻(2-溴乙氧基)苯基-10,15,20-三(对甲氧基苯基)卟啉(2)参照文献[6]方法合成。
加入5-氟尿嘧啶130 mg(1 mmol)和 N, N'-二甲基甲酰胺(DMF)30 mL于100 mL干燥圆底烧瓶中,加热搅拌,促进5-氟尿嘧啶溶解。 再加入焙烧过的无水K2CO3 166 mg(1.2 mmol ),油浴80 ℃电磁搅拌反应1 h,使5-氟尿嘧啶生成钾盐,然后向其中加入100 mg(0.12 mmol)5-邻(2-溴乙氧基)苯基-10,15,20-三(4-甲氧基苯基)卟啉(2),升温至120 ℃,10 h后停止反应。 冷却,用饱和NaCl溶液盐析,抽滤,水洗,干燥得紫色晶体。 粗产品用柱色谱分离,用氯仿与丙酮体积比为10:1的混合液淋洗,收集第二色带,旋转蒸发浓缩,干燥得17 mg紫色晶体化合物3,产率为16%。
称取化合物3 49.6 mg(0.056 mmol)溶于20 mL二氯甲烷中,加入110.8 mg(0.56 mmol) 氯化锰溶于DMF制成的饱和溶液,电磁搅拌下回流,反应2 h。 冷却,水洗,分液。水浴蒸干。 粗产品用氯仿与丙酮体积比为5:1的混合液洗脱。 收集最浓绿色带,旋转蒸发蒸去溶剂,干燥得绿色固体3a 41.8 mg,产率为80.4%。 按上述方法,用102.9 mg(0.56 mmol )乙酸锌代替氯化锰,得紫红色固体3b 35.7 mg,产率为67.9%。
初步筛选:将传代培养的肺腺癌细胞株A549、肝癌细胞株Bel7402和人结肠癌细胞株HCT-8,用胰酶消化后,用含体积分数10%小牛血清的RPMI-1640 培养液配成浓度为1.5×104个/mL 的细胞悬液。 接种于96孔培养板内,每孔接种180 μL(含2700个肿瘤细胞),37 ℃下5%CO2环境培养24 h。 实验组加样品20 μL,每种样品平行3个孔,每孔终体积为200 μL,用1640培养液补足,使得样品终浓度为10 mg/L,另设阳性对照组。 置于37 ℃条件下5%CO2环境培养48 h。 后弃上层清液,每孔加入100 μL新鲜配制的0.5 g/L MTT 无血清培养液( 用RPMI-1640 配制,避光保存于-20 ℃)。 37 ℃继续培养4 h,小心弃去上层清液,并加入150 μL二甲基亚砜(DMSO)溶解甲臜(formazon)沉淀,用微型超声振荡器混匀,在酶标仪上测定波长570 nm处的光密度值(OD)。 以无药肿瘤细胞培养为对照组,计算药物对肿瘤细胞的抑制率:
肿瘤细胞的生长抑制率/%=
在此基础上,进一步筛选:样品抑制率大于50%的样品进行半数抑制率(IC50)实验,即将待测样品以0.08、0.4、2、10和50 mg/L不同浓度加入96孔培养板内,每个浓度平行3个孔,实验方法同上,测定波长570 nm处的光密度值(OD),通过Excel中的Forecast计算IC50值。
化合物3:UV-Vis(CHCl3), λmax/nm:Soret band 421.4,Q band 517.5,553.8,592.3,648.8;IR(KBr), σ/cm-1:3473.80(N—H),2922.16,2850.79(C—H、—CH2—),1637.56(C=O in 5-FU),1506.41,1465.90(benzene ring and porphyrin ring C=C),1379.10(C—N),1246.02(C—O—C);1H NMR(CDCl3,400 MHz), δ:2.714(s,2H,porphyrin N—H),4.082(m,9H+2H,3OCH3+OCH2),4.309~4.322(m,2H,—NCH2),7.289~7.421(m,6H+4H,3Ar- m-H+ArO- o, m, p-H),7.759(s,1H,C—H in 5-FU),7.997~8.230(m,6H,3Ar- o-H),8.683~8.860(m,8H,pyrrole-H)。
化合物3a:UV-Vis(CHCl3), λmax/nm:Soret band 480.4,Q band 585.0,622.0;IR(KBr), σ/cm-1:3423.65(N—H),2920.23(C—H,—CH2—),1637.56(C=O in 5-FU),1415.15,1492.90(benzene ring and porphyrin ring C=C),1380.10(C—N),1248.02(C—O—C),1006.84(Mn—N)。
化合物3b:UV-Vis(CHCl3), λmax/nm:Soret band 422.6,Q band 549.8,589.4;IR(KBr), σ/cm-1:3448.72(N—H),2920.23,2850.79(C—H、—CH2—),1653.00(C=O in 5-FU),1458.18,1516.05,1568.13(benzene ring and porphyrin ring C=C),1381.03(C—N),1244.09(C—O—C),995.27(Zn—N);1H NMR(CDCl3,400 MHz), δ:4.025~4.069(m,9H+2H,3OCH3+ OCH2),4.314~4.328(m,2H,—NCH2),7.208~7.385(m,6H+4H,3Ar- m-H+ArO- o, m, p-H),7.745(s,1H,C—H in 5-FU),7.923~8.191(m,6H,3Ar- o-H),8.732~8.937(m,8H,pyrrole-H)。
从上述紫外可见光谱数据可以看出:1)化合物3的紫外光谱有一个Soret带和4个Q带。 根据Gouterman的四轨道模型[7],在421.4 nm处的吸收带是Soret带,由a1μ( π)-eg( π*)电子跃迁产生,归属为卟啉环内 π-π*跃迁的第二电子激发态;吸收光谱在500~650 nm之间的是Q带,由a2μ( π)-eg( π*)电子跃迁产生,归属为卟啉环内 π-π*跃迁的第一电子激发态;2)化合物3配合金属离子形成化合物3a和3b后,紫外光谱中QI、QⅣ带消失。 分析原因是,Zn2+、Mn2+是 d10组态的金属离子,当与自由卟啉配位后,形成N—M键,配合物的对称性发生变化,且能级靠近,分子轨道的分裂程度减少,简并度增加,Q带数目相应减少,表现为QI、QⅣ谱带消失[8];3)与化合物3b相比,化合物3a的Soret带明显红移至480.4 nm。 分析原因为:两种金属离子的相对离子半径大小顺序为Mn2+(88 pm)>Zn2+(74 pm),而电负性Mn2+(1.5)<Zn2+(1.6),因此锰对外层价电子的束缚能力较弱,使得电子更容易流向卟啉环,从而引起整个卟啉环的电子云密度升高,降低卟啉环的电子跃迁所需要的能量,使得Soret带红移较多,因此,化合物3a具有锰卟啉的结构特征[9]。
从上述红外光谱数据可以看出:在化合物3中,羰基的特征吸收峰出现在1637.56 cm-1,在1246.02 cm-1处有醚键的典型特征吸收峰[10],表明5-FU是以醚键的形式与卟啉侧端的乙氧链相连。 再观察化合物3a和3b的红外光谱数据,分别在1006.84和995.27 cm-1处出现了新的强伸缩振动吸收峰,该峰归属于N—Mn键和N—Zn键的特征吸收峰,这些数据均表明,合成了稳定的金属卟啉配合物。
参照文献[6]对化合物3和3b的核磁共振氢谱进行了归属,由上述数据可以看出:化合物3中卟啉环内N—H键的化学位移在 δ -2.714处,这是由于卟啉环的大环π电子体系,在外磁场作用下,会产生一个对抗的感应磁场,而卟啉环内的NH处于很强的屏蔽区,因此NH的化学位移向高场移动,移到了参考峰TMS( δ 0.00)的高场,故 δ为负值。 而在化合物3b中该峰消失,说明生成了新的金属卟啉配合物;而5-FU中的N—H由于没有处在屏蔽区,故它的 δ为正,应在9以上[10]。 但由于受溶剂的影响,积分比又小,该峰未显示出来。
分别测试了化合物3、3a和3b对肺腺癌细胞株A549、肝癌细胞株Bel7402和人结肠癌细胞株HCT-8的抑制活性,抑制率见表1。 结果表明:1)样品浓度为10 mg/L时,除化合物3对A549抑制率为负值外,3种样品对上述3种细胞株均有一定的抑制作用。 但抑制活性均低于阳性对照药5-氟尿嘧啶,也未达到文献[5]的数值,分析原因可能是卟啉-5-FU化合物对细胞株有很强的选择性,对上述3种细胞株选择性较差,对其它细胞株的抑制活性将在后续工作中进行;2)卟啉配体3a对3种细胞株的抑制率明显高于化合物3和3b,抑制率分别为50.15%、55.35%和31.27%,说明卟啉化合物的抗肿瘤活性与中心金属离子有关,锰卟啉对癌细胞的抑制活性较锌卟啉和自由卟啉要高很多;3)化合物3a对人结肠癌细胞株HCT-8的抑制率最高,为55.35%,也表明卟啉化合物的抗肿瘤活性具有一定的选择性,对不同的肿瘤细胞的作用机理可能不一样[11]。 此外,对抑制率大于50%的化合物3a进行半数抑制率IC50实验,结果表明,锰卟啉对人结肠癌细胞株HCT-8的半抑制浓度IC50为17.8 mg/L,具有一定的细胞毒作用。 有关取代基的不同以及碳链长短对抗肿瘤活性的影响正在研究中。
| 表1 化合物3、3a、3b对A549、Bel7402、HCT-8的抑制率 Table 1 Inhibition rates of compound 3, 3a and 3b to A549, Bel7402 and HCT-8 |
合成了一种5-氟尿嘧啶修饰的自由卟啉及其2种金属卟啉配合物:锰卟啉和锌卟啉,并对3种化合物结构和性能进行了表征。 与自由卟啉和锌卟啉相比,锰卟啉对HCT-8肿瘤细胞的半抑制浓度IC50为17.8 mg/L,具有较好的抗肿瘤活性,在人结肠癌治疗领域具有潜在的应用前景。
致谢:抑制活性由中国医学科学院药物研究所国家药物筛选中心测试,特此感谢。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|