
MAI Wenpeng, YANG Liu, LYU Mingxiu, et al. Metal-Free Synthesis of 3,3-Difluoro-2-Oxindoles[J]. Chinese Journal of Applied Chemistry, 33(11): 1279-1283


共同通讯联系人:卢奎,教授; Tel:0371-62509959; E-mail:lukui126@126.com; 研究方向:多肽合成
发展了一种合成3,3-二氟-2-氧化吲哚及其衍生物的新方法。 以简单的芳香胺和溴二氟乙酸乙酯为起始原料,在无溶剂和N2气保护下搅拌5 h,得到含二氟的氮乙酰苯胺衍生物中间体,产率85%~93%。 该中间体与碘甲烷反应,得到 N-甲基保护的乙酰苯胺衍生物,产率75%~90%。 该衍生物在3.0化学计量促进剂次硫酸氢钠甲醛的存在下,以 N, N-二甲基甲酰胺/H2O为溶剂,发生分子内自由基关环反应,得到3,3-二氟-2-氧化吲哚及其衍生物,产率53%~72%。 该方法原料便宜,不使用任何金属化合物,最后一步以水相为溶剂,较为环保,具有潜在的工业应用价值。
Co-corresponding author:LU Kui, professor; Tel:0371-62509959; E-mail:lukui126@126.com; Research interests:polypeptide synthesis
A novel method for the synthesis of 3,3-difluoro-2-oxindoles was developed. 2-Bromo-2,2-difluoro- N-phenylacetamide was prepared from neat simple aromatic amines with ethyl bromodifluoroacetate. Reflux of the product in CH3CN with CH3I for 24 h gave 2-bromo-2,2-difluoro- N-methyl- N-phenylacetamide in 75%~90% yields. In the presence of 3.0 chemometric number sodium formaldehyde sulfoxylate, this key intermediate undergoes intramolecular radical cyclization in aqueous solution( N, N-dimethylformamide/H2O) and affords the final product 3,3-difluoro-1-methylindolin-2-ones in 53%~72% yields. This method can be performed in aqueous solution from commercially available and affordable starting materials without any metal catalysts. It shows potential in industrial production of 3,3-difluoro-2-oxindoles.
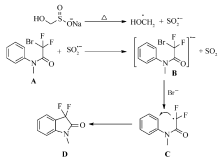
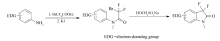

近年来,含氟化合物的合成及其方法学研究已经成为有机合成中研究热点之一,特别在药物研发领域。 原因是有机小分子接上氟原子后,可以提高其亲油性,若作为药物则可提高其新陈代谢稳定性和生物相容性[1,2];在材料学方面,氟原子的引入,也可以改变材料的特性,使其具有独特的性质,如材料的透气性等[3]。
氧化吲哚的衍生物广泛存在于天然产物和生物活性物质中,例如含有3-氧化吲哚结构的靛蓝染料[4]。 近5年来,3,3-二取代-2-氧化吲哚的合成引起了研究人员广泛的兴趣,许多新合成方法被报道出来[5,6,7,8,9,10]。 其核心结构氧化吲哚具有潜在的药用价值,已用于多种抗菌剂和抗癌药中,如盐酸罗匹尼罗,一种含有氧化吲哚结构多巴胺激动剂。 然而,很少有报道将氟原子直接引入到氧化吲哚的3位,而是均将含氟基团接入到氧化吲哚结构外。 将氟原子的特性和氧化吲哚的药用价值结合起来有很大的研究空间,氧化吲哚的生物利用性和其药物活性有可能发生显著变化,因此,合成3,3-二氟-2-氧化吲哚有重要的意义。
目前,合成3,3-二氟-2-氧化吲哚的方法仅有两个。 2010年,胡金波等[11]报道了一种以铜为促进剂合成上述化合物的方法。 该方法使用摩尔分数为200%的铜粉作为促进剂,以碘代芳香酰胺化合物为底物,二甲基亚砜(DMSO)为溶剂,在65 ℃下反应7 h,产率在31%~61%。 其后,该方法没有得到改进和发展,直到2015年,Buchwald等[12]报道了一个以钯为催化剂的分子内关环反应合成3,3-二氟-2-氧化吲哚及其衍生物的方法。 该方法使用摩尔分数为2%的钯盐,摩尔分数为8%的特制膦配体,K2CO3作碱,在120 ℃下反应20 h,产率在52%~91%。 上述方法中,贵金属作催化剂和昂贵的膦配体的使用使后处理过程繁琐,合成成本较高,限制了这两种方法的进一步应用。
在我们的前期研究中发现[13],次硫酸氢钠甲醛可以引发带有吸电子基团的卤代烃发生单电子转移(SET),进而生成自由基,发生分子间的自由基加成反应。 本文进一步将该方法应用到分子内的自由基关环反应上(Scheme 1),优化了反应条件,探讨了可能的反应机理。
 | Scheme 1 Synthsis of 3,3-difluoro-2-oxindole |
RE2000B型旋转蒸发仪(巩义予华仪器设备有限公司),Bruker Avance 400型核磁共振仪(德国Bruker公司),CDCl3为溶剂,四甲基硅烷(TMS)为内标;Q-Tof MS/MS型高分辨质谱(美国Waters公司)。
溴二氟乙酸乙酯(纯度>98%,北京百灵威科技有限公司),次硫酸氢钠甲醛(纯度>98%,上海TCI试剂有限公司)。 芳香胺、碘甲烷、碳酸钾、乙腈、 N, N-二甲基甲酰胺(DMF)购自上海阿拉丁试剂有限公司,均为分析纯。
芳香胺(0.91 mL,10 mmol),溴二氟乙酸乙酯(1.92 mL,15 mmol),加入到100 mL的单口圆底烧瓶中,N2保护后,在25 ℃下,搅拌6 h后,加入30 mL水和50 mL乙酸乙酯,再搅拌5 min,将该溶液转入到分液漏斗中,收集有机层,乙酸乙酯层先后用稀盐酸水溶液(0.5 mol/L)和饱和食盐水各洗涤1次,无水硫酸钠干燥,在45 ℃下减压蒸出溶剂,得到淡黄色粉末2-溴-2,2-二氟- N-(芳香基)乙酰胺2.72 g,产率为90%~97%。
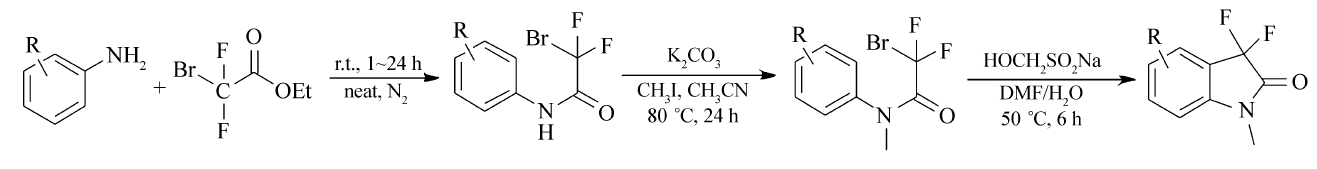 | Scheme 2 Metal-free preparation of 3,3-difluoro-2-oxindoles |
2-溴-2,2-二氟- N-(芳香基)乙酰胺(1.25 g,5 mmol)和碘甲烷(0.93 mL,15 mmol)溶解于15 mL的乙腈溶剂中,加入1.38 g无水K2CO3作碱,在80 ℃下回流反应24 h,冷却至室温,过滤除去不溶性盐,减压蒸去溶剂,剩余物中加乙酸乙酯溶解,然后用饱和食盐水洗涤,干燥,柱层析分离,洗脱剂为 V(石油醚): V(乙酸乙酯)=4:1,纯化后即得到2-溴-2,2-二氟- N-(芳香基)- N-甲基乙酰胺。
2-溴-2,2-二氟- N-(芳香基)- N-甲基乙酰胺(0.26 g,1.0 mmol和次硫酸氢钠甲醛(0.46 g,3.0 mmol)分别加入到一个50 mL的圆底烧瓶中,然后依次分别加入2 mL DMF和2 mL H2O, 在50 ℃下搅拌反应6 h,反应完毕,冷却至室温,加入20 mL乙酸乙酯,混合液一起转入到分液漏斗中,用饱和食盐水20 mL洗涤3次,有机层用无水硫酸钠干燥1 h,柱层析分离,洗脱剂为 V(石油醚): V(乙酸乙酯)=4:1,得到3,3-二氟-1-甲基吲哚啉-2-酮(目标产物),核磁鉴定产物的纯度可达95%。
3,3-二氟-5-甲氧基-1-甲基吲哚啉-2-酮:白色固体,mp 82~84 ℃,产率72%。1H NMR(400 MHz, CDCl3 ), δ:7.15(dd,1H, J=2.0,4.0 Hz),7.02~7.05(m,1H),6.84(dt,1H, J=1.2,8.8 Hz ),3.85(s,1H,OCH3),3.22(s,1H,NCH3);13C NMR(100 MHz,CDCl3), δ:165.1(t, J=31 Hz,C=O),156.7,137.0,121.2(t, J=22 Hz),118.4,111.1,110.3,55.8,26.4;19F NMR(376 MHz,CDCl3), δ:-112.2;HRMS计算值C10H10F2NO2[M+H]+ 214.0674,实测值214.0672。
3,3-二氟-5-甲基-1-甲基吲哚啉-2-酮:透明液体,产率为62%。1H NMR(400 MHz,CDCl3 ), δ:7.39(s,1H),7.30~7.32(m,1H),6.82(d, J=8.0 Hz,1H),3.22(s,1H,NCH3),2.39(s,3H);13C NMR(100 MHz,CDCl3), δ:165.3(t, J=30 Hz, C=O ),141.5,133.8,125.3,120.1(t, J=23 Hz),113.6,111.1,109.2,26.3,20.9;19F NMR(376 MHz,CDCl3), δ:-112.2;HRMS计算值C10H10F2NO[M+H]+ 198.0725,实测值198.0728。
3,3-二氟-1-甲基吲哚啉-2-酮:淡黄色固体,mp 55~57 ℃,产率为70%。1H NMR(400 MHz,CDCl3), δ:7.51~7.58(m,2H),7.18~7.22(m,1H),6.91(d,1H, J=8.0 Hz),3.24(s,3H,NCH3);13C NMR(100 MHz,CDCl3), δ:165.27(t, J=30 Hz, C=O),143.94,133.66,124.55,123.91,120.01(t, J=22 Hz),110.91(t, J=247 Hz),109.54,26.26;19F NMR(376 MHz,CDCl3), δ:-112.36;HRMS计算值C9H8F2NO[M+H]+ 184.0568,实测值184.0566。
3,3,5-三氟-1-甲基吲哚啉-2-酮:白色固体,mp 47~48 ℃,产率为53%。1H NMR(400 MHz,CDCl3), δ:7.31~7.34(m,1H),7.21~7.23(m,1H),6.86~6.89(m,1H),3.241(s,3H,NCH3);13C NMR(100 MHz,CDCl3), δ:165.00(t, J=30 Hz, C=O),160.67,158.24,139.87,120.13,113.04,110.53,26.45;19F NMR(376 MHz,CDCl3), δ:-112.46,-117.57;HRMS计算值C9H7F3NO[M+H]+ 202.0474,实测值202.0471。
5-溴-3,3-二氟-1-甲基吲哚啉-2-2酮:淡黄色固体,mp 58~60 ℃,产率为68%。1H NMR(400 MHz,CDCl3), δ:7.65~7.66(m,1H),7.59~7.63(m,1H),6.83(d, J=8 Hz,1H),3.22(s,3H);13C NMR(100 MHz,CDCl3), δ:165.33(t, J=30 Hz, C=O),143.50,136.41,133.30,129.06,127.87,111.13,26.43;19F NMR(376 MHz,CDCl3 ), δ:-112.27;HRMS计算值C9H7BrF2NO[M+H]+ 261.9674,实测值261.9668。
以甲氧基苯胺为起始底物,合成2-溴-2,2-二氟- N-(4-甲氧基苯基)乙酰胺,然后考察了反应促进剂用量、反应温度、反应溶剂对关环反应的影响,结果见表1。
| 表1 反应条件优化 a Table 1 Optimization of reaction conditions a |
以次硫酸氢钠甲醛(1.0化学计量)为促进剂,乙腈和水混合溶液作为反应溶剂,50 ℃下,关环反应发生,产率仅为35%。 增加促进剂用量为1.5化学计量,反应温度为80 ℃,反应产率略微增加至43%。 更换溶剂,如二氧六环/水、四氢呋喃/水、丙酮/水;即使促进剂的量增加至3.0化学计量,反应也没有发生,说明溶剂对该反应有着重要的影响。 将溶剂更换为溶解能力更强的DMF/H2O为反应液,反应在50 ℃下进行,3.0化学计量的次硫酸氢钠甲醛为促进剂,反应顺利进行,产率达到72%。 进一步增加次硫酸氢钠甲醛的用量,或是增加反应温度至80 ℃,反应产率反而下降。 为了进一步筛选最佳的反应条件,将混合溶剂中的水变为1.0 mol/L的盐酸水溶液或1.0 mol/L的碳酸氢钠水溶液(表1,entry 9 and 10 ),结果反应仍可发生,但只得到中等产率。
{kind=link}
{kind=link}
{kind=link}
本文提供了一种无金属参与的合成3,3-二氟-2-氧化吲哚的新方法。 廉价的、商业化的次硫酸氢钠甲醛为促进剂,可在水相中(DMF/H2O)引发自由基反应,实现分子内关环,产率在70%左右。 与现有的合成此类化合物的两种合成方法相比,该方法避免了铜粉和贵金属Pd的使用,免除了繁琐的清除残留金属的后处理步骤,最后一步主要在水相中进行,较为环保,合成成本低,但该方法只适用于苯环上带有推电子基团(EDG)的芳香胺,而带有吸电子基团的芳香胺由于电子云密度低导致分子内关环反应不能发生。此方法具有潜在的工业应用价值。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|