

XIA Ran, SUN Liping, QU Guirong, et al. Synthesis of Anti-leukemia Drug Cladribine[J]. Chinese Journal of Applied Chemistry, 33(11): 1274-1278


报道了抗白血病药物克拉屈滨的合成新方法:在NaH作用下,廉价易得的6-氯嘌呤高选择性地在 β位和1-氯-2-脱氧-3,5-二- O-对氯苯甲酰基- D-核糖缩合; β-缩合物的2位在三氟乙酸酐和四丁基硝酸铵作用下引入硝基;在NH4Cl/EtOH作用下,2-硝基转化为2-氯;最后在饱和NH3/CH3OH溶液中完成保护基脱除和6-氯氨解两步反应,以4步共43.5%的总收率得到抗白血病药物克拉屈滨。 该方法完全避免了 α异构体的生成,原料廉价易得,分离纯化不需柱层析,且反应扩大到100 g规模时,收率无下降,具有较好的应用前景。
Cladribine, an anti-leukemia drug, was synthesized in 43.5% total yield by 4 steps. Readily available 6-chloropurine was glycosylated with 1-chloro-3,5-di- O-p-benzoyl- D-ribose with good selectivity for β anomer in the presence of NaH. The C2-H of β-glycosylated product was converted into C2-NO2 using 2,2,2-trifluoroacetic anhydride and tetrabutylammonium nitrate, followed by the transformation of C2-NO2 into C2-Cl in NH4Cl/EtOH solution. The deprotection step and ammonolysis of C6-Cl were accomplished in NH3/CH3OH. The separation of α-anomer, expensive starting materials and chromatography are not required. Moreover, cladribine could be produced on a 100 gram scale with maintained yield, indicating good potential in industrial applications.
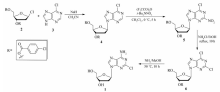
克拉屈滨(cladribine,化合物1),化学名2-氯-2'-脱氧腺嘌呤核苷,1993年首次在美国上市,商品名Leustatin,由Onho Biotech和Johnson&Johnson公司共同研发,用于治疗毛细胞白血病(HCL)和Walden-Strom氏巨球蛋白血病[1]。 作为2'-脱氧核苷类代表性药物[2],是一线的核苷类抗白血病药物,国内外需求量大。
文献报道的克拉屈滨的合成方法主要有以商品化核苷为原料的碱基[3,4,5]、糖基[6]修饰法,以及以嘌呤碱基和脱氧核糖衍生物为原料的缩合法[7,8,9],后者以其原料廉价易得更受到关注。 缩合法的不足之处是缩合时产生近一半的 α异构体,难以分离,收率降低。 如以2,6-二氯嘌呤和脱氧核糖为原料[7],收率仅8%。 Robins等[8]将咪唑基团引入嘌呤6位,然后再和1-氯-2-脱氧-3,5-二- O-对氯苯甲酰基- D-核糖缩合,因咪唑基团的位阻较大,会减少 α异构体生成,收率可以达到59%,但引入和脱除咪唑基增加了反应步骤。 岑均达等[9]将2-氯腺嘌呤和1-氯-2-脱氧-3,5-二- O-对氯苯甲酰基 -D-核糖缩合,收率可达69%,但2-氯腺嘌呤的量是1-氯-2-脱氧-3,5-二- O-对氯苯甲酰基 -D-核糖的2.3倍,成本较高。 故开发高效、 β选择性好的克拉屈滨的合成方法具有重要意义。
蒋忠良等[10]曾报道以NaH为碱,乙腈为反应溶剂,6-氯嘌呤和1-氯-2-脱氧-3,5-二- O-对氯苯甲酰基 -D-核糖缩合,可以高选择性地得到 β-缩合物6-氯-2'-脱氧-3',5'-二- O-对氯苯甲酰基- β-D-呋喃基嘌呤核苷。 受此启发,我们在缩合物2位引入硝基,继而将2-硝基转化为2-氯[11],最后将6位氯原子氨解,以4步和43.5%的总收率得到克拉屈滨。 避免了 α异构体的生成,原料廉价易得,且反应规模扩大到100 g时,收率无下降,具有较好的应用前景(Scheme 1所示)。
 | Scheme 1 The synthetic route of cladribien(compoud 1) |
{kind=link}
AC 400型核磁共振仪(德国Bruker公司),DMSO-d6或CDCl3为溶剂,TMS为内标;Q-TofMS/MS型高分辨质谱仪(美国Waters公司);XRC-1型显微熔点仪(四川大学科仪厂),温度计未校正;CJF-0.25型高压反应罐(巩义市予华仪器有限公司)。
6-氯嘌呤、1-氯-2-脱氧-3,5-二- O-对氯苯甲酰基 -D-核糖(纯度>99%,新乡拓新生化股份有限公司)。 所用试剂均为市售分析纯。 乙腈加入硫酸镁干燥后减压蒸馏处理,CH2Cl2用4Å分子筛干燥处理24 h。 其它试剂未经进一步处理。
1.2.1 6-氯-2'-脱氧-3',5'-二- O-对氯苯甲酰基 -β-D-呋喃基嘌呤核苷的合成(化合物4) 根据文献[10]方法制备,收率72%,产物为淡黄色油状物,1H NMR数据与文献[10]报道相符。1H NMR(CDCl3,400 MHz), δ:8.57(s,1H),8.24(s,1H),8.16~8.13(m,4H),7.50~7.47(m,4H),6.60~6.56(m,1H),5.86(t, J=4.4 Hz,1H),4.80(ddd, J1=3.6 Hz, J2=12.4 Hz, J3=15.6 Hz,2H),4.68(q, J=3.6 Hz,1H),3.27~3.20(m,1H),2.87~2.58(m,1H);13C NMR(CDCl3,100 MHz), δ:166.2,165.9,155.8,153.2,149.6,138.6,133.7,129.8,129.6,128.6,84.8,82.9,75.3,37.8,18.4。
1.2.2 6-氯-2-硝基-2'-脱氧-3',5'-二- O-(对氯苯甲酰基)- β-D-呋喃基嘌呤核苷的合成(化合物5) 四丁基硝酸铵(10.1 g,33 mmol)溶解于无水CH2Cl2(50 mL)中,冷却至0 ℃,加入三氟乙酸酐(4.6 mL,33 mmol),搅拌20 min,加入新制备的化合物4(12.1 g,22 mmol),保持0 ℃反应5 h,加入饱和Na2CO3溶液(100 mL),搅拌,分出有机相,水相用CH2Cl2(20 mL)萃取两次,合并有机相,无水Na2SO4(1 g)干燥,减压(真空度≈0.011 MPa)除去溶剂,得到淡黄色油状物10.56 g,即化合物5,收率81%。1H NMR(CDCl3,400 MHz), δ:8.28(s,1H),8.01~7.97(m,4H),7.47~7.45(m,4H),6.45~6.41(m,1H),5.62~5.60(m,1H),4.72(ddd, J1=3.6 Hz, J2=12.4 Hz, J3=15.6 Hz,2H),4.52(q, J=3.6 Hz,1H),2.73~2.68(m,1H),2.36~2.28(m,1H);13C NMR(CDCl3,100 MHz), δ:165.32,163.3,156.2,151.9,149.0,139.9,131.2,130.9,129.2,129.0,119.3,85.0,82.4,75.1,64.4,37.8,12.4;HRMS计算值C24H17Cl3N5O7[M+H]+ 592.0188,实测值592.0184。
1.2.3 2,6-二氯-2'-脱氧-3',5'-二- O-(对氯苯甲酰基)- β-D-呋喃基嘌呤核苷的合成(化合物6) 化合物5(5.9 g,10 mmol)加入到无水乙醇(20 mL)中,加入NH4Cl(0.8 g,15 mmol),加热回流反应10 h,减压(真空度≈0.011 MPa)除去溶剂,无水乙醇重结晶,得到白色固体6,4.77 g,收率82%。 mp 210~212 ℃。1H NMR(DMSO-d6,400 MHz), δ:8.25(s,1H),8.00(t, J=4.4 Hz,4H),7.63~7.58(m,4H),6.28(t, J=6.8 Hz,1H),5.63~5.60(m,1H),4.60(ddd, J1=4.4 Hz, J2=12.0 Hz, J3=16 Hz,2H),4.49(t, J=4.0 Hz,1H),2.62~2.50(m,2H);13C NMR(DMSO-d6,100 MHz), δ:164.7,164.5,163.6,150.4,138.6,135.8,131.2,131.1,129.0,128.9,110.0,84.3,80.8,74.9,35.8,11.9;HRMS计算值C24H16Cl4N4NaO5[M+Na]+ 602.9767,实测值602.9765。
1.2.4 克拉屈滨的合成(化合物1) 化合物6(5.82 g,10 mmol)加入到预先装有饱和NH3/CH3OH溶液(200 mL)的高压反应釜中,加热到50 ℃(压力约1.2 MPa),反应10 h,降温,释放压力,反应液减压(真空度≈0.011 MPa)除去溶剂,用水重结晶,得到白色固体克拉屈滨2.60 g,收率91%。 白色固体,mp 306 ~ 310 ℃(dec.)(文献[9]mp >300 ℃)。1H NMR(DMSO-d6,400 MHz), δ:8.34(s,1H),7.34(brs,2H),6.34(dd, J1=6.0 Hz, J2=10.6 Hz,1H),5.34(d, J=18.8 Hz,1H),4.11(t, J=2.4 Hz,1H),3.88(q, J=4.0 Hz,1H),3.57(dd, J1=11.2 Hz, J2=3.6 Hz,2H),3.76~3.69(m,1H),2.28~2.23(m,1H);13C NMR(DMSO-d6,100 MHz), δ:155.9,152.2,148.7,139.4,119.0,87.8,83.8,70.8,61.7,28.4;HRMS计算值C10H13ClN5O3[M+H]+286.0701,实测值286.0703。
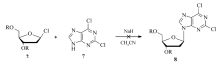
如果提前引入氯原子,即使用2,6-二氯嘌呤而不是6-氯嘌呤,在和1-氯-2-脱氧-3,5-二- O-对氯苯甲酰基- D-核糖缩合时, β选择性差,不能高收率地得到带保护基的 β-缩合物;而6-氯嘌呤和1-氯-2-脱氧-3,5-二- O-对氯苯甲酰基- D-核糖缩合时, β异构体的收率可以达到72%,所以6-氯嘌呤是最佳选择。
 | Scheme 2 The condensation of 2,6-dichloropurine(compound 7) with 2-deoxy-3,5-di- O-( p-chlorobenzoyl)- D-ribofuranosyl chloride(compound 2) |
{kind=link}
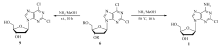
在室温条件下,化合物6和饱和NH3/CH3OH溶液反应,得到2'-脱氧-2,6-二氯嘌呤核苷(化合物9,收率65%)。 随着反应温度的提高,化合物9的比例逐渐下降,化合物1的比例逐渐增加。 经过优化,在50 ℃反应10 h,化合物1的收率最高,无化合物9的生成(Scheme 3,表1)。 继续升高温度,收率稍有提高,但是较高的温度使反应釜压力增大,操作不安全。 综合考虑,以50 ℃反应10 h为宜。反应结果也显示,反应顺序为:脱酰基>6-氯氨解,可以通过控制反应温度和反应时间,得到相应的产物。 这与文献[4]报道的结果一致。 本路线也为目标产物的类似物的合成提供了一个便捷的方法。
 | Scheme 3 The effects of temperature on the ammonolysis of compound 6 |
{kind=link}
| 表1 氨解反应条件的优化 a Table 1 The optimization of ammonolysis a |
为了增加该反应的实用性,我们还考察了关键步骤硝化和氯代的反应规模对收率的影响(表2)。 结果显示,在适当延长反应时间后,硝化步骤可以扩大到100 g,氯代步骤可以扩大到200 g,收率没有下降,分离纯化不需柱层析,操作及后处理均非常简便。
| 表2 不同反应规模对收率的影响 a Table 2 The effects of reaction scales on the yield a |
由表2可知,将反应规模从10 g级扩大到100 g级,经过常规的分离提纯后,硝化收率可以达到80%。 在反应规模达到200 g时,氟代步骤的收率可以达到83%。 对母液回收利用,收率还会有所增加,所以,总体上整条路线的收率高,分离纯化简便,应用性强。
以廉价的6-氯嘌呤和1-氯-2-脱氧-3,5-二- O-对氯苯甲酰基 -D-核糖为原料,高选择性地缩合得到 β异构体后,通过硝化/氯代反应,在嘌呤碱基的2位引入氯原子,6位氯原子在饱和的NH3/CH3OH溶液氨解,以4步和43.5%的总收率得到抗白血病药物克拉屈滨。 该方法完全避免了 α异构体的生成,原料廉价易得,分离纯化不需柱层析,操作简便,且反应规模扩大到100 g规模时,收率无下降。 为克拉屈滨的合成提供了一条新的合成途径,具有潜在的应用前景。
辅助材料(Supporting Information)[中间体及产物1H NMR和13C NMR图谱]可以免费从本刊网站(
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|