
WANG Dongsheng, LI Wentao, YANG Xiaofang, et al. Ferrates:Green Oxidants and Coagulants in Water Treatment[J]. Chinese Journal of Applied Chemistry, 33(11): 1221-1233


多种新型污染物和微生物污染等问题的出现,导致地表水水质复杂多变,传统的水处理药剂和处理方式已无法满足人们对饮用水处理的需求。 高铁酸盐作为一种新型水处理试剂,同时具备优良的氧化性和混凝性,而且不会引起二次污染,是一种可大力开发的绿色试剂。 本文综述了高铁酸盐净水剂的制备与表征分析方法,及其用于水处理对重金属、新型污染物和微生物等去除的作用机制。 目前,有关高铁酸盐用于有机污染物去除的混凝和氧化去除协同作用的研究尚不多见,高铁酸盐的氧化-混凝协同特性尚未被充分开发。 本文以此为重点进行了讨论,并对高铁酸盐净水剂的应用进行了展望。
The rise of emerging contaminants and microorganisms causes the complexity of drinking water quality and brings a gap between peoples demand and water treatment efficiency using conventional treatment reagents and techniques. Ferrate is an effective and multi-functional green water purification material, which shows both good oxidation and coagulation ability without secondary pollution. This paper reviews the removal mechanism of contaminants including heavy metal ions, emerging contaminants and microorganisms by ferrate. At present, the investigation of ferrates oxidation and coagulation cooperative effect is insufficient and the application of ferrates in water treatment has not been fully developed. Therefore, the oxidation and coagulation cooperative effect of ferrates is emphatically discussed to direct the application of ferrates in water treatment. Finally, the prospect of application of ferrates in water treatment is commented.







据统计,全球60亿人口中的20%人口面临着饮用水缺乏的问题。 另外,饮用水水源污染日益严重,其中30%人口使用的水源存在微生物污染,0.14%人口使用的水源存在砷污染[1]。 预计到2025年,大约三分之二的人口将处于饮用水缺乏的境地[2]。 饮用水问题已成为21世纪最大的难题之一,亟需高效和可持续的水处理技术来满足人类日益增长的饮用水需求。 铁基材料被广泛应用于水处理过程中,如零价铁原位修复地下水、芬顿反应和磁性分离等[3,4,5],主要是因为铁元素无毒无污染,处理效果优良,并且铁元素在地球分布极广,价格低廉。
铁元素有多种价态,包括零价、+Ⅱ、+Ⅲ、+Ⅳ、+Ⅴ和+Ⅵ等。 其中,高价铁备受关注,常用于工业合成或水处理,如绿色化学合成、有机污染物的降解等。 高铁酸盐(FeⅥ)具有强氧化性,可以在数秒到数十秒内降解一些难降解有机物(如天然有机物、内分泌干扰物等),而且不会带来二次污染[6,7,8]。 Sharma等[6]报道了在中性条件下,磺胺类物质被高铁酸盐氧化时对应的半衰期在十几秒到数十秒之间,表明在极短时间内高铁酸盐能将磺胺类物质氧化分解。 与高锰酸钾相比,其氧化性更高,无重金属污染;与次氯酸盐相比,高铁酸盐不会产生氯代消毒副产物[6];与臭氧相比,高铁酸盐不会与溴离子反应生成具有潜在致癌能力的溴酸盐[7,8]。 同时,高铁酸盐对碘离子的氧化速率远低于次氯酸盐与臭氧,速率低4~6个数量级[8,9]。 除此之外,高铁酸盐可以混凝去除水体中的胶体颗粒物。 与传统无机铁盐(如硫酸亚铁、硝酸铁)相比,高铁酸盐对水体浊度去除效率高,1 min内就可以使胶体颗粒脱稳[10],并且能够去除一些有毒的重金属离子,如Hg2+、Pb2+、Cr3+和Cd2+等[11]。
本文综述了高铁酸盐(FeⅥ)的制备与表征方法及其氧化混凝作用的机制,主要讨论在水处理中高铁酸盐去除重金属、难降解有机物和微生物的研究进展,并对氧化和混凝的协同作用做了初步探讨。 通常情况下,高铁酸盐被有机物还原或自身分解后转变为三价铁化合物,化合物的形态很大程度受周围环境因素的影响,将直接决定后续的混凝效果,需要系统深入的研究。
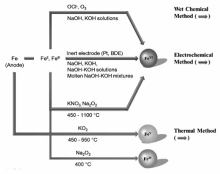
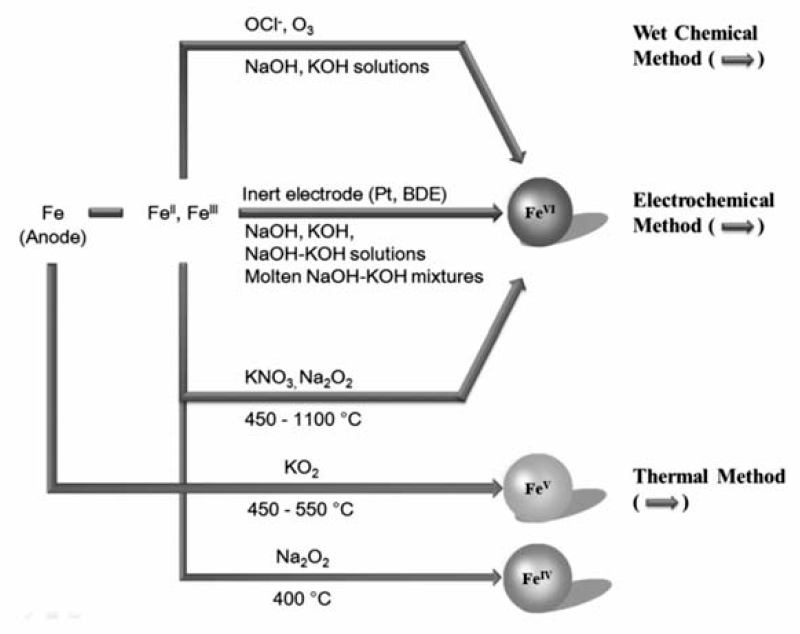
常用的高铁酸盐制备方法有3种,即:湿式氧化法、电化学氧化法和高温氧化法(如图1所示[12])。 湿式氧化法是在高浓度NaOH条件下,利用次氯酸盐氧化铁盐(硝酸铁、氯化铁)形成高铁酸钠,然后利用高铁酸钾溶解度低于高铁酸钠的特性,向高铁酸钠溶液中加入KOH,析出高铁酸钾晶体。 该方法制备的产物产率和纯度高,但是工艺复杂,是实验室常用的制备方法。 也有文献[13]报道,用臭氧代替次氯酸盐制备高铁酸盐,方便快捷但是产率较低。 电化学氧化法是一个相对比较清洁的方法,采用铁单质或化合物(零价铁、亚铁盐、铁盐等)作为电极,在14 mol/L NaOH溶液中电氧化制备高铁酸盐,提升温度可以增加氧化效率,但同时也会促进高铁酸盐(Ⅵ)的分解,为解决这个问题,可以采用Pt或掺杂B的材料做阳极,熔融态碱做电解质,以减少阳极对高铁酸盐合成的影响[14]。 高温氧化法是将铁氧化物和硝酸钾混合,然后加热到1100 ℃煅烧,形成高铁酸钾,但是纯度很低(~30%)。 为了提高纯度,用过氧化钠替换硝酸钾,在600 ℃下煅烧,可获得纯度为90%高铁酸盐固体[15]。 此外,该方法常用于制备高铁酸盐(Ⅳ),在370 ℃下煅烧氧化铁与过氧化钠的混合物( n(Fe): n(Na)=1:2)即可获得。
高铁酸盐有多种浓度分析的方法,如亚铬酸盐法和亚砷酸盐氧化滴定法等,但最常用的是分光光度法。 高铁酸盐呈紫黑色,在510 nm处有很强的吸收峰(最大摩尔吸收系数为1150 L/(mol·cm)),在275与320 nm处有两个肩峰,通常根据510 nm处的吸光度计算高铁酸盐的摩尔浓度。 考虑到高铁酸根存在分解的可能性,研究者[13,16]提出了更为准确的方法:1)利用碘离子(I-)与高铁酸根反应,即:Fe
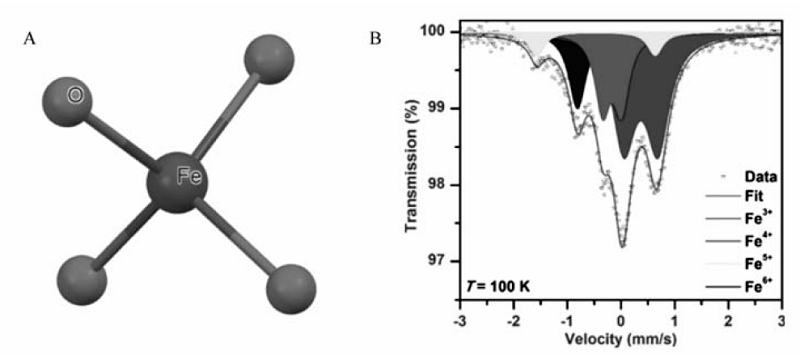
高铁酸根具有四面体结构,其中Fe原子位于四面体的中心,4个O原子位于四面体的4个顶角,具有Td对称性,如图2(A)所示,4个Fe—O键键长在自由状态下长度相同,O—Fe—O夹角为109°28',二者在受晶体场环境影响时会有变化,在K2FeO4晶体中,Fe—O键长在0.166~0.167 nm范围内,和K2MnO4差不多,比K2CrO4略长[17]。 常见的高铁酸盐的表征方法有红外光谱(FTIR)、X射线衍射(XRD)、X射线光电子能谱(XPS)和X射线吸收精细结构(EXAFS)。 高铁酸盐的红外特征峰出现在750~900 cm-1,是由Fe—O键振动和四面体结构变化引起的。 XPS研究表明,高铁酸根的Fe2 p3/2电子结合能出现在712 eV左右,而含铁酸根(Ⅴ和Ⅳ)的Fe2 p3/2电子结合能出现在711 eV左右。 上述表征方法基本都是辅助手段,通常受其它因素干扰很严重。 穆斯堡尔谱被认为是表征高铁酸盐中铁形态最有效的手段,如图2(B)所示,高铁酸盐在自身分解过程中,同质异能位移( δ)逐渐增大,说明铁价态发生了变化,有低价态的Fe形成,拟合分析得知,中间出现了五价、四价铁,最后转化为三价铁[12]。
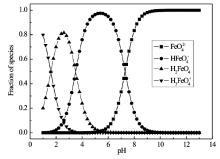
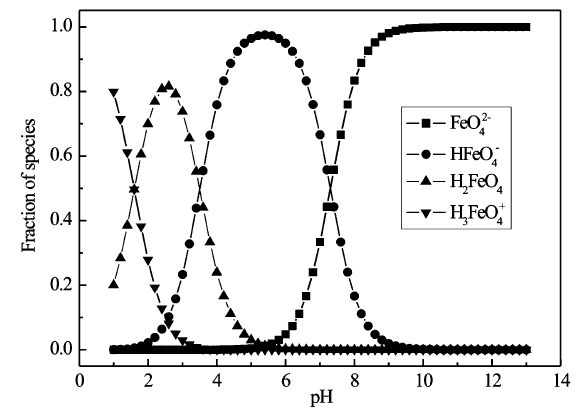
如表1所示,高铁酸盐在酸性与碱性条件下的氧化还原电势分别为2.20和0.70 V。 酸性环境下,高铁酸盐的氧化性显著高于其它氧化剂,酸性条件下氧化还原电势依次是:高铁酸盐>臭氧>双氧水>高锰酸盐>次氯酸>溶解氧,碱性条件下氧化还原电势依次是:臭氧>双氧水>次氯酸盐>高铁酸盐>高锰酸盐>溶解氧。 高铁酸盐是一种强力的氧化剂,在干燥环境中较为稳定,但在酸性水溶液中极其不稳定,高铁酸盐会迅速被水还原为三价铁化合物,同时释放氧气。 随着pH值升高,高铁酸盐的稳定性逐渐增加,pH值在11.5~13.5范围内分解较慢[18]。 造成这种结果的原因,可以由H+与Fe
| 表1 常见氧化剂的标准电极电势 Table 1 Standard electrode potential of common oxidants |
研究人员发现[21],有机物被高铁酸盐氧化后的产物,不仅与自身结构和所含基团有关,还与高铁酸根的浓度有关。 脂肪烃类化合物可被高铁酸盐氧化为羟基化合物[22];醇类化合物被氧化为为酮类和醛类[23,24];甲胺类化合物被氧化为甲酰胺或甲酸类化合物[25];酚类化合物被氧化为苯醌类或双酚类化合物[26,27];含硫的脂肪化合物或硫醇类化合物则被氧化为磺酸化合物或硫酸根[22,28]。 高铁酸盐过量时,苯胺类化合物被氧化为硝基苯,反之则为偶氮苯[29,30]。
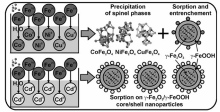
高铁酸盐在反应过程中表现出良好的混凝和吸附特性,可用于去除水体中的重金属离子。 Lim等[31]研究了高铁酸盐对Cu2+、Mn2+和Zn2+3种离子的去除效果,当Fe和M2+的摩尔比分别为1:1、1:1和1:3时,其去除率达到最大,分别为100%、70%和100%,当天然有机物(NOM)在体系中存在时,可提高Cu2+和Mn2+的去除率。 Prucek等[32]报道了高铁酸盐对Cu2+、Ni2+、Co2+、Cd2+和Al3+离子的去除研究,发现高铁酸盐对它们的去除能力如下:Al3+>Cu2+>Co2+>Ni2+>Cd2+,并且去除效率随着pH值的升高而增大,这主要是由高铁酸盐氧化后,形成的纳米羟基铁或铁氧化物对离子进行吸附和包裹引起的。 他们还对高铁酸盐原位去除游离态重金属离子的机制做了研究(图4),提出铁氧化物晶格结构置换(M2Fe2O4)和表面吸附(>FeO-M)机制是重离子去除的主导过程,机制原理与重金属离子的自身特性(如离子半径等)密切相关。
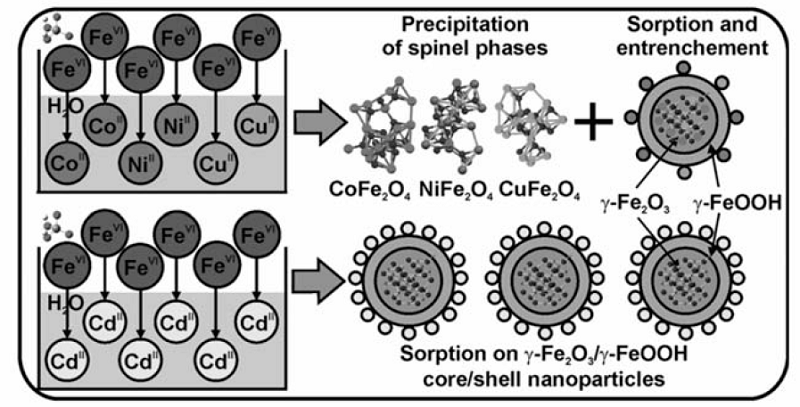
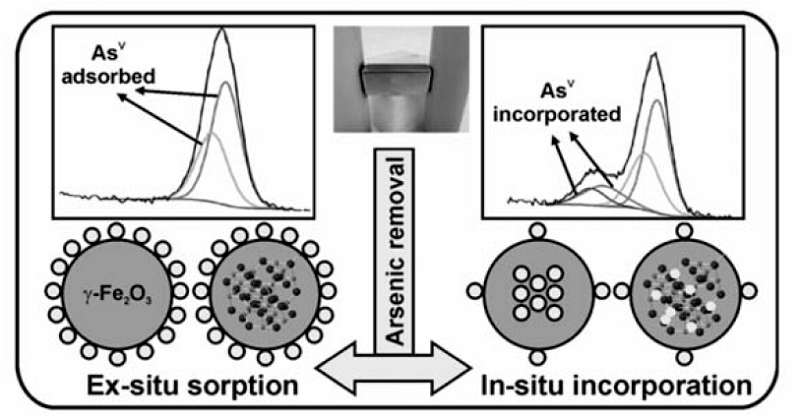
高铁酸盐对亚砷酸盐也有很好的处理效果。 Robert等[33]研究发现,高铁酸盐与亚砷酸盐作用时,当Fe/As质量比设定为5,亚砷酸根几乎可全部被去除。 另有研究[34]表明,某些金属离子能促进高铁酸盐对亚砷酸盐的去除效果,如在Al3+或Fe3+存在的条件下,用少量高铁酸盐即可完全去除亚砷酸盐(低于1 μg/L),而磷酸根、NOM和硅酸根等离子的存在,却可抑制高铁酸盐对亚砷酸根的去除,可能是由它们同亚砷酸根竞争高铁酸根导致的。图5为高铁酸盐原位去除亚砷酸盐的机制:首先亚砷酸盐被氧化为砷酸盐,然后部分砷酸盐被铁氧化物包裹于内部,余下部分则被吸附于铁氧化物表面,从而达到去除效果。 这些猜测由57Fe穆斯堡尔谱与高分辨X射线能谱所证实。 由于复杂的水质因素(如pH值、温度、NOM和重金属离子种类等)直接影响高铁酸盐的反应与混凝特性,进而会影响到重金属的去除效率。 对于水体中NOM和磷酸根等如何影响高铁酸盐去除重金属离子,需做进一步研究。
新型有机污染物,属于持久性难降解有机污染物,如内分泌干扰物、药物及个人护理品等,疏水性很强,存在于水体环境和沉积物中,虽然含量很低(ng/L~μg/L),但产生的危害极大,对人类健康有着潜在的威胁。 去除这一类有机物,常见的处理方法是高级氧化法,如臭氧或次氯酸盐氧化等,但这些氧化剂用于水处理后会引起一系列的二次污染。 例如,氯气处理会产生带有毒性的氯代副产物;二氧化氯与臭氧的使用分别会产生亚氯酸盐和溴酸盐,进一步形成有毒的副产物;而用氯胺处理则会产生亚硝胺类污染物[35]。 与上述氧化剂相比,高铁酸盐则不会产生有毒性的副产物,高效且对环境友好。
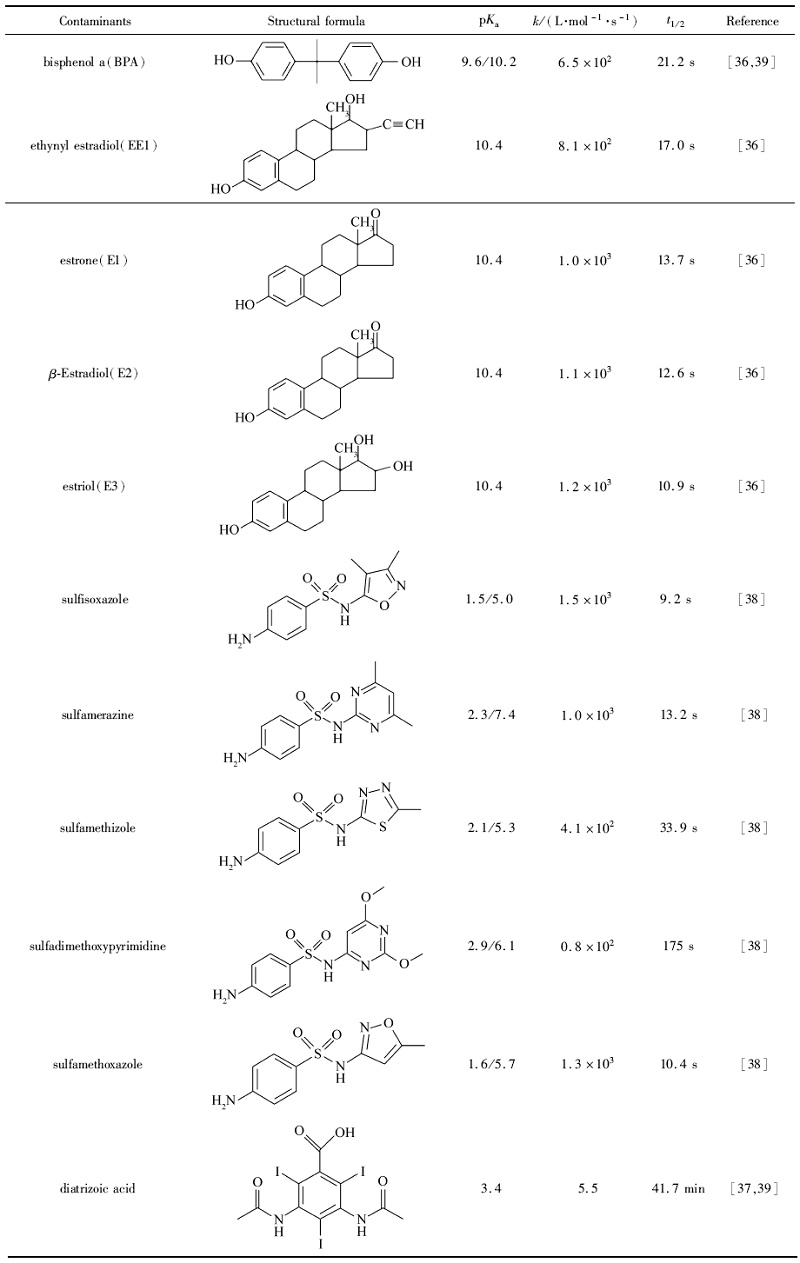
高铁酸盐氧化降解新型有机污染物的速率,与污染物的自身特性密切有关。如表2所示,有机污染物的分解速率常数从5.5×103~1.5×103 L/(mol·s)均有分布,有机物的结构和所含基团种类与数量均会影响氧化降解的速率。通过投加过量的高铁酸盐(10 mg/L),可以推断出污染物降解的半衰期,数秒到数分钟,少量有机物的半衰期以分钟计算,如磺胺地索心、泛影酸等,表明这些污染物需要更长的时间来降解,不易去除。
| 表2 部分新型有机污染物与高铁酸盐(Ⅵ)的二级化学反应速率常数(pH=7.0,25 ℃) Table 2 Pesudo-second-order rate constants of parts of organic contaminants by ferrate(Ⅵ) (pH=7.0,25 ℃) |
有学者对高铁酸盐氧化降解酚类化合物的反应动力学过程做了研究,如双酚A、壬基酚和四溴苯酚等[36,37]。 研究发现,当高铁酸盐与双酚A的摩尔比达到5:1时,双酚A可以被完全去除,其溶解性有机碳(DOC)去除率达80%。 当高铁酸盐不足时,DOC去除率则降低,降解不完全的产物残存于溶液中。 当采用液相-质谱联用技术对其进行分析时,发现了9种中间产物,根据检测到的中间产物,推测可能的反应机制如图6所示。 首先,双酚A被高铁酸盐氧化为苯酚、4-异丙醇基-苯酚、4-异丙烯基-苯酚和1-苯基-1-丁烯基-苯,这4种化合物进一步被氧化为苯乙烯、4-异丙基-环己酮-2,5-二烯酮、4-乙酰基-苯酚、草酸和丙二酸,最后经过C—C键断裂,转化为苯醌、对二苯酚、苯乙烯、苯酚和小相对分子质量的有机酸。

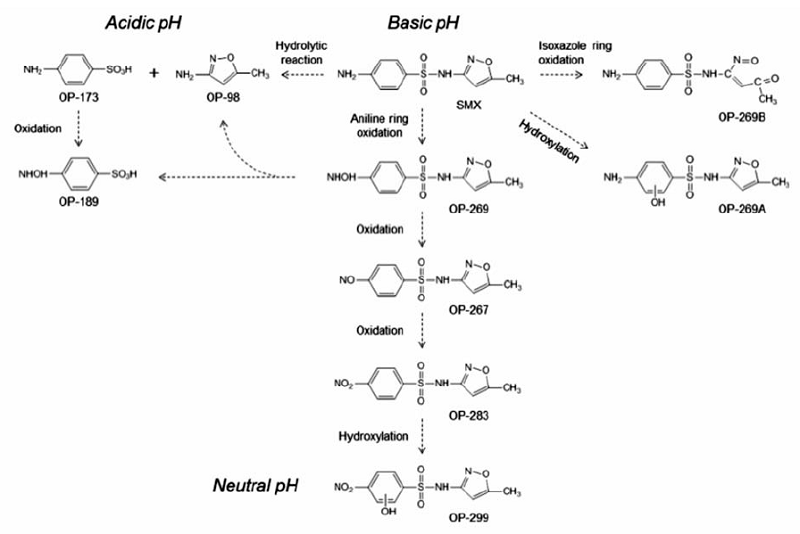
抗生素存在于药品和个人护理产品中,由于其难降解且具有生物传递性,对周围环境产生很大的污染,已引起学者广泛的关注,其中磺胺类药品的研究较多。 Kim等[40]对高铁酸盐氧化降解磺胺甲恶唑做了研究,发现当高铁酸盐与磺胺甲恶唑的摩尔比为4:1时,可有效去除磺胺甲恶唑。 氧化过程中,磺胺甲恶唑的S—N键断裂,苯环发生羟基化、芳胺氧化和异恶唑开环反应后,转化为含有3-氨基-5甲基异唑、羟胺基、亚硝基和硝基基团的磺胺甲恶唑衍生物。 经色谱技术分析,研究者提出了磺胺甲恶唑的氧化降解途径,如图7所示。 可以看出,在不同pH值条件下,高铁酸盐氧化降解磺胺甲恶唑的途径也不同。
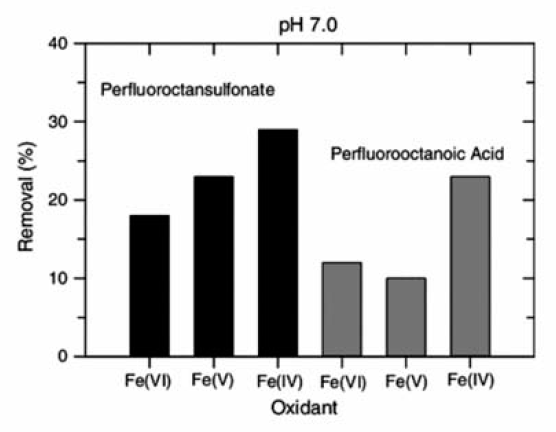
高铁酸盐氧化具有化合物和条件选择性,Brain等[41]研究了高价态的高铁酸盐(Fe(Ⅳ)/Fe(Ⅴ)/Fe(Ⅵ))氧化降解全氟化合物的反应,发现在pH=9条件下,Fe(Ⅵ)对全氟辛酸和全氟辛烷基磺酸基本没有去除,而Fe(Ⅴ)与Fe(Ⅳ)对二者有很好的去除效果,全氟辛烷基磺酸的去除率分别达20%、35%,全氟辛酸的去除率均在15%左右(图8)。 而在pH值7的条件下,Fe(Ⅵ)的氧化去除效果增加,如图9所示,去除率达到17%和12%左右,而Fe(Ⅳ)和Fe(Ⅴ)的去除率变化则不明显或下降,这表明Fe(Ⅵ)的氧化性对pH值的依赖度很高,同时,化合物的类型直接决定了氧化去除的结果。
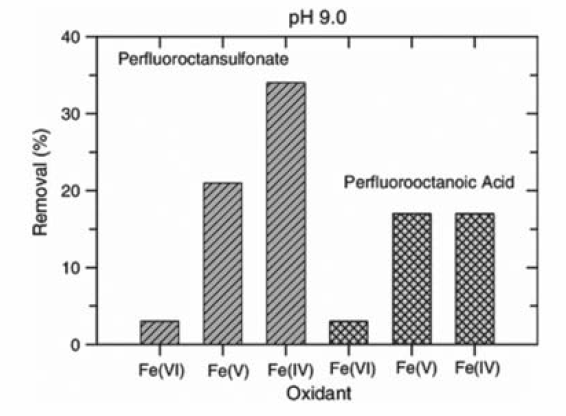 | 图8 pH=9时Fe(Ⅳ)/Fe(Ⅴ)/Fe(Ⅵ)对全氟化合物的去除率[41]Fig.8 Removal efficiency of perfluorinated compounds via ferrate(Ⅵ/V/Ⅳ) at pH=9[41] |
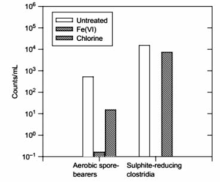
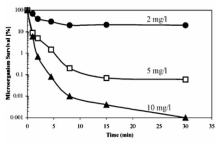
高铁酸盐可用作水处理消毒剂,以杀灭细菌和病毒,比如杆菌、球菌等[42]。 研究发现,用高铁酸盐( ρ(Fe)=6 mg/L)作为消毒剂,可在7 min内杀死99.9%以上的大肠杆菌[43]。 同时,高铁酸盐能使抗氯细菌和硫酸盐还原菌失活[44],与次氯酸钠( ρ(Cl)=3.5 mg/L)相比,高铁酸盐( ρ(Fe)=2 mg/L)使好氧孢子失活的数目高出2个数量级(如图10)[44]。 另外,研究发现高铁酸盐对MS2噬菌体、f2病毒等病毒有一定的抑制性[45,46]。 Schink等[47]发现病毒失活时间与pH值成反比关系,如:pH=6.9 耗时5.7 min,pH=5.9耗时0.77 min。 当pH=7.8时,高铁酸盐(Ⅵ)的投加量增到 ρ(Fe)=10 mg/L, f2病毒去除率达到99.9%,如图11所示。
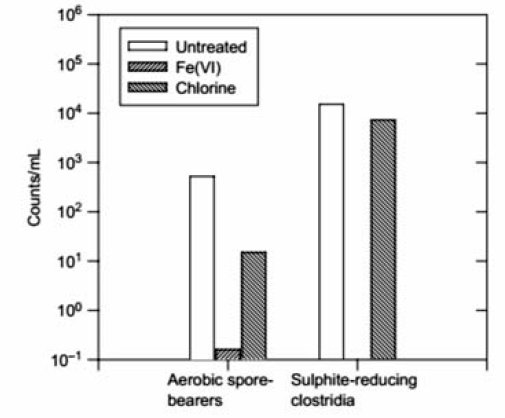 | 图10 30 min内细菌的失活数量[44]Fig.10 Disinfection of river water by Fe(Ⅵ) over 30 min contact time[44] sodium ferrate(Ⅵ): ρ(Fe)=2 mg/L; hypochlorite: ρ(Cl)=23.5 mg/L |
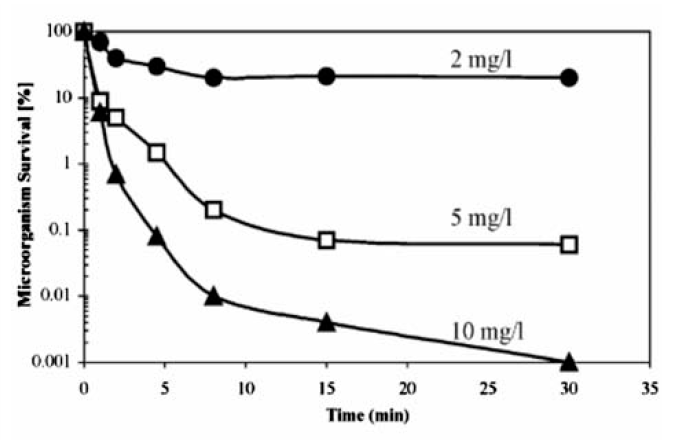 | 图11 不同浓度高铁酸盐(Ⅵ)对F2病毒失活率的影响 (pH=7.8,24 ℃)[47]Fig.11 Effect of different Ferrate concentration on the inactivation of F2 virus(pH=7.8,24 ℃)[47] |
与其它消毒剂相比,高铁酸盐去除微生物的效率更高,且对水体没有二次污染。高铁酸盐(Ⅵ)使微生物失活的机制是依靠高铁酸盐强大的氧化能力破坏蛋白质结构,使其变性。
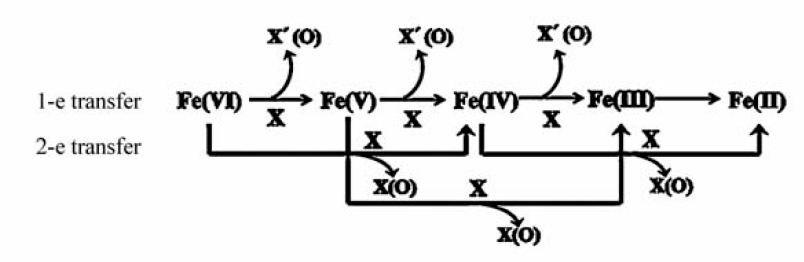
高铁酸盐氧化去除污染物的作用机制,主要依据化学计量数和中间产物推断出可能的反应过程,并利用二级动力学反应速率方程进行模拟论证。 如图12所示,其机制主要包括:1)高铁酸盐经过1e或2e传输过程被还原为Fe(Ⅳ)或Fe(Ⅴ)化合物,污染物被氧化为某些活性物种;2)Fe(Ⅳ)或Fe(Ⅴ)化合物继续氧化降解有机污染物,同时Fe(Ⅵ/V/Ⅳ)发生歧化反应,最终形成铁氧化物。
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
污染物种类不同,其与高铁酸盐作用的氧化机制也有所不同。 高铁酸盐在氧化氰化物和硫化物等无机物时,以1e(Fe(Ⅵ)→Fe(Ⅴ)→Fe(Ⅳ)→Fe(Ⅲ))方式转移电子进行反应,1 mol高铁酸盐Fe(Ⅵ)会得到3 mol电子[48,49];而在氧化含氧酸根(如亚砷酸根、亚硫酸根和亚硝酸根等)时,则以2e(Fe(Ⅵ)→Fe(Ⅳ)→Fe(Ⅱ))方式转移电子进行反应,1 mol Fe(Ⅵ)会得到4 mol电子,最后Fe(Ⅳ)或Fe(Ⅵ)与Fe(Ⅱ)反应生成Fe(Ⅲ)[34,50,51]。 在高铁酸盐氧化有机物过程中,1 mol Fe(Ⅵ)可以氧化3 mol抗坏血酸分子(Asc)[52],即:Fe(Ⅵ)/Fe(Ⅴ)/Fe(Ⅳ)+3Asc→Fe(Ⅲ)+3Asc·-;在氧化半胱氨酸时,1 mol Fe(Ⅵ)得到4 mol电子还原为Fe(Ⅱ)[53];在氧化蛋氨酸时,反应中1 mol Fe(Ⅵ)得到3 mol电子被还原为Fe(Ⅲ)[54];而在氧化2,2'-联氮双(3-乙基苯并噻唑啉-6-磺酸)二铵盐(ABTS)时,却仅发生了1 mol的电子转移[21]。
综上所述,高铁酸盐在氧化降解污染物过程中,Fe(Ⅵ)可以被还原到Fe(Ⅲ)和Fe(Ⅱ)两种价态,其电子转移途径与污染物种类有很大关系,尤其是在降解有机污染物时,1e与2e电子转移方式均可能存在。 根据前面的论述,污染物分子在与Fe
高铁酸盐发生氧化反应后,Fe(Ⅵ)转化为Fe(Ⅲ)形成羟基铁氧化物(或铁氧化物),呈现出混凝特性。 Fe(Ⅵ)在去除重金属离子时,以羟基铁氧化物内部包裹(如M2Fe2O4)与表面吸附为主。 而在去除水体胶体和颗粒物时,絮凝效果通常优于传统的无机混凝剂[55,56],因为在高铁酸根被还原的过程中,会经历从Fe(Ⅵ)到Fe(Ⅲ)的梯段变价演变过程,生成一系列正价态中间化合物,这些化合物可能具有比较丰富的网状结构,在压缩颗粒物的扩散层过程中,同时起到电中和、吸附和网捕的效果,强化了凝聚特性,表现出独特的絮凝效果[57]。
在发挥高铁酸盐氧化性的同时,如何将之后的混凝特性加以应用,最大限度地发挥高铁去除污染物的能力,同时减少反应中的副产物,是应用高铁酸盐的最好前提。 由于Fe3+与有机物通常可以发生较强的络合作用,当Fe与有机物分子的摩尔比较高时,Fe3+则可以对有机分子产生很强的混凝作用,反之则会形成可溶的配合物,所以控制高铁酸盐的投加量很重要。同时研究发现,环境因素(pH值、磷酸根、腐殖酸和金属离子等)对高铁酸盐分解后产生絮体粒径产生重要影响,中性条件下,少量磷酸盐或腐殖酸的存在,可以有效增大高铁酸盐分解后产生的絮体粒径,提高了污染物的去除效果。 对于高铁酸盐的氧化效果,发现加入金属离子,可以加速有机污染物的去除。 以上实验现象表明,控制好环境条件,可促使高铁酸盐最大程度地发挥氧化和混凝作用,在精确控制反应要素的条件下,可以达到氧化与混凝协同作用的结果,以最大限度的去除污染物。
高铁酸盐作为一种绿色的水处理剂,最大的优势在于高效的氧化和混凝处理效果,同时不产生二次污染。 高铁酸盐与水体中卤素离子的反应速率很低,几乎可以忽略,这样不会产生含卤素毒害物。 在处理重金属离子时,重金属离子以嵌入铁氧化物晶格内或被吸附于铁氧化物表面的方式去除,后续处理时不会被浸出产生污染。 高铁酸盐在氧化降解过程中,通过1e或2e电子转移的方式反应,Fe(Ⅵ)最终被还原为Fe(Ⅲ)或Fe(Ⅱ),具体的反应途径取决于污染物的特性。
将高铁酸盐用于水处理具有巨大的应用价值,然而在实际工程操作和工艺处理时,高铁酸盐的生产和其稳定特性,限制了其大规模的实际应用。 由于缺乏更为系统且深入的研究,很多问题需要进一步的探索,存在的诸多不足如下:1)实验室中制备高铁酸盐的技术比较成熟,但是由于操作复杂、成本高,无法满足规模化的工业生产,需对实验方法作进一步改进更新;2)污染物降解途径与反应条件、污染物形态密切相关,如弱酸性条件(HFe
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|
| [26] |
|
| [27] |
|
| [28] |
|
| [29] |
|
| [30] |
|
| [31] |
|
| [32] |
|
| [33] |
|
| [34] |
|
| [35] |
|
| [36] |
|
| [37] |
|
| [38] |
|
| [39] |
|
| [40] |
|
| [41] |
|
| [42] |
|
| [43] |
|
| [44] |
|
| [45] |
|
| [46] |
|
| [47] |
|
| [48] |
|
| [49] |
|
| [50] |
|
| [51] |
|
| [52] |
|
| [53] |
|
| [54] |
|
| [55] |
|
| [56] |
|
| [57] |
|