
ZHANG Dandan, ZHA Xianyu, GAO Baojiao. Synthesizing and Bonding Bidentate Schiff Base Ligand on Side Chain of Polysulfone and Preliminary Exploration of Luminescence Property of Functionalized Polysulfone-Tb(Ⅲ) Complexes[J]. Chinese Journal of Applied Chemistry, 33(1): 53-62


使氯甲基化聚砜(CMPSF)的氯甲基与乙醛酸(GA)的羧基发生酯化反应,将乙醛(AL)基团键合在聚砜侧链,制得改性聚砜PSF-AL;接着,又使3-氨基吡啶(AP)的伯氨基与PSF-AL的醛基发生席夫碱反应,在聚砜侧链合成与键合了具有特定结构的双齿席夫碱配基AA,从而获得了双齿席夫碱配基功能化聚砜PSF-AA,采用红外光谱(FTIR)和核磁共振氢谱(1H NMR)等技术手段对其结构进行了表征。 以功能化聚砜PSF-AA为大分子配体,与Tb(Ⅲ)离子及Eu(Ⅲ)离子分别配位,制得了二元高分子-稀土配合物PSF-(AA)3-Tb(Ⅲ)和PSF-(AA)3-Eu(Ⅲ),初步探索了两种配合物的光致发光性能。 重点研究了功能化聚砜PSF-AA的制备反应,考察与分析了主要因素对CMPSF与GA之间酯化反应的影响规律,优化了反应条件。 CMPSF与GA之间的酯化反应属氯烷的亲核取代反应,实验结果表明,适宜的溶剂为极性较强的 N, N-二甲基乙酰胺,75 ℃为适宜的反应温度。 大分子配体PSF-AA对Eu(Ⅲ)离子不产生敏化作用,而对Tb(Ⅲ)离子的荧光发射则产生强烈的敏化作用,配合物PSF-(AA)3-Tb(Ⅲ)发射出较强的Tb(Ⅲ)离子的特征荧光,即表现出发射绿光的光致发光性能。
The esterification of chloromethylated polysulfone(CMPSF) with glyoxylic acid(GA) afforded aldehyde(AL) modified PSF-AL. Subsequent condensation of PSF-AL with 3-aminopyridine led to bidentate Schiff base ligand-functionalized PSF-AA. The chemical structure of PSF-AA was characterized by FTIR and1H NMR spectra. The binary polymer-rare earth complexes of PSF-(AA)3-Tb(Ⅲ) and PSF-(AA)3-Eu(Ⅲ) were prepared and their photoluminescence properties were preliminarily explored. Optimized esterification between CMPSF and GA were performed in N, N-dimethylacetamide at 75 ℃. The macromolecular PSF-AA does not exhibit sensitization towards Eu(Ⅲ) ion, whereas PSF-AA can strongly sensitize the fluorescence emission of Tb(Ⅲ) ion. The complex PSF-(AA)3-Tb(Ⅲ) emits characteristic green fluorescence of Tb(Ⅲ) ion.



在发光材料领域,键合型高分子-稀土配合物是一类十分重要的材料,在光致发光、电致发光、光电通讯、激光以及太阳能转换系统等领域,该类发光材料都有着重要的应用价值和发展前景[1,2,3,4,5]。 该类材料不但保留了小分子稀土配合物独特而优异的发光性能,还兼具有高分子化合物优良的力学性能和易于加工成型的特点(特别是易于成膜的特性)[6,7],而且与小分子稀土配合物掺杂于聚合物基质所形成的发光材料相比,键合型高分子-稀土配合物发光材料呈现内部均相的特质,这些特性都十分有利于其实际应用。 目前,较成功的键合型高分子-稀土配合物发光材料的研究报道还较少[8,9],需要大力发展,以满足高新科学技术的需求。
制备键合型高分子-稀土配合物有两种基本的方式,或通过含可聚合双键的稀土配合物的聚合反应而获得[10],或通过聚合物大分子链上的配基与稀土离子配位而形成[11]。 制备含稀土配合物的单体往往比较困难,且聚合所得产物的相对分子质量受限;而通过大分子反应先将小分子配基键合在聚合物侧链(或原聚合物侧链上就含有配基)形成大分子配体,而后再使之与稀土离子配位形成键合型高分子-稀土配合物,是比较易于进行的一种方法。 然而,在已报道的键合型高分子-稀土配合物发光材料体系中,聚合物侧链中参与配位的配基多数为脂肪羧基(主要来自于单体丙烯酸或甲基丙烯酸),脂肪羧基对稀土离子只起配位螯合的作用,无敏化功能(即无“Antenna” 效应),需要加入协同配位的小分子配体,才能形成发光材料[12,13],这在很大程度上局限了该类功能材料的发光性能。 针对此种情况,本课题组通过大量的研究实践,提出了一个重要的新思路:设法在聚合物侧链键合对稀土离子兼具有配位螯合和敏化发光双重功能的配基,是制备高发光性能的高分子-稀土配合物材料的有效途径。 在前期的研究中,我们将具有此双功能的芳羧酸(苯甲酸和萘甲酸)配基引入聚砜及聚苯乙烯侧链,键合的配基对稀土离子既可进行螯合配位,形成稳定的高分子-稀土配合物,又能大幅度地发生分子内能量转移,强烈地敏化稀土离子的荧光发射,制得了多种高性能的光致发光高分子-稀土配合物及其薄膜[14,15,16,17]。
席夫碱化合物也是一类重要的配基,它们不但能与过渡金属及稀土金属离子发生配位作用,而且还能以多齿配基的形式存在,与金属离子实现螯合配位,从而形成稳定的配合物;更重要的是席夫碱配基大多具有共轭芳环,因此具有很强的光吸收性能,可能会发生强烈的分子内能量转移,因此,多齿的席夫碱配基也是一类兼具有配位螯合和敏化发光双重功能的配基。 基于这一推断,在前期的研究中,我们将苯甲醛基团键合于聚砜侧链,制得了双齿席夫碱配基功能化的聚砜,通过大分子配体与Eu(Ⅲ)离子的配位,首次获得了发射红光的席夫碱型高分子-稀土配合物[18]。 本文则通过分子设计的另一思路,设法将乙醛基团键合于聚砜侧链,制得了另一种结构的双齿席夫碱配基功能化的聚砜,以该功能化的聚砜为大分子配体,与Tb(Ⅲ)离子配位,成功地获得了发射绿光的席夫碱型高分子-稀土配合物。本文重点研究大分子配体的制备与表征,初步探索了所制备的双齿席夫碱配基功能化聚砜与不同稀土离子形成的配合物的发光性能,而对它们发光性能与发光机理以及结构与性能关系更深入的研究结果,将另文报道。 本文设计制备了具有一定结构的双齿席夫碱配基功能化的聚砜,从而制得了发射绿光的席夫碱型高分子-稀土配合物。显然,本文的研究结果在光致发光高分子-稀土配合物的设计与制备方面具有重要的参考价值。
聚砜(PSF,
1700型傅里叶红外光谱仪(FTIR,美国Perkin-Elmer公司);UV-2602型紫外/可见分光光度计(上海尤尼柯公司);DRX300型核磁共振仪(瑞士Bruker公司);HITACHIF-2500荧光光度计(日本日立公司)。
1.2.1 双齿席夫碱配基功能化聚砜PSF-AA的制备 通过三步大分子反应,将双齿席夫碱配基键合在聚砜侧链,制得双齿席夫碱配基功能化聚砜。
1)参照文献[19]所述步骤,制备氯甲基化聚砜(CMPSF)。 以自制的BCMB为氯甲基化试剂,SnCl4为路易斯酸催化剂,制得CMPSF,测得其氯含量为1.8 mmol/g。
2)通过CMPSF氯甲基的酯化反应,将乙醛基团键合于聚砜侧链。 在装有电动搅拌器、冷凝回流管的四口瓶中,加入0.5 g CMPSF和50 mL溶剂DMAC,使CMPSF充分溶解,再加入0.233 g乙醛酸GA(3.15 mmol)和1.90 g缚酸剂Na2CO3(1.8 mmol),恒温于75 ℃,在搅拌条件下,使GA与CMPSF之间的酯化反应进行12 h。 反应结束后,加入沉淀剂乙醇,将产物沉出。 抽滤,用乙醇和蒸馏水反复洗涤,真空干燥至恒重,即得侧链键合乙醛(AL)基团的改性聚砜PSF-AL。 采用氧弹燃烧—佛尔哈德滴定法测定其氯含量,计算出CMPSF大分子链中氯甲基的转化率,它标志着改性聚砜PSF-AL分子链中乙醛基团AL的键合量大小。
3)通过PSF-AL的席夫碱反应,将双齿席夫碱配基键合于聚砜侧链。 在四口瓶中,加入0.5 g改性聚砜PSF-AL和 50 mL 溶剂DMAC,完全溶解后,加入0.075 g的3-氨基吡啶AP,恒温于70 ℃,在N2气氛围及搅拌条件下,使PSF-AL和AP之间的席夫碱反应进行8 h。 反应结束后,加入150 mL沉淀剂乙醇,静置后取上清液(上清液体积共200 mL),待测定。 经过滤,分离收集产物聚合物,用乙醇和蒸馏水交替洗涤,真空干燥至恒重,即制得侧链键合有双齿席夫碱配基的功能化聚砜PSF-AA(因双齿席夫碱配基是键合AL和小分子AP反应的结果,故将此功能化聚砜指定为PSF-AA)。
1.2.2 功能化聚砜PSF-AA的表征 1)KBr 压片法测定聚砜PSF、氯甲基化聚砜CMPSF、改性聚砜PSF-AL和产物PSF-AA的红外光谱,通过比较,确定功能化聚砜PSF-AA的化学结构;2)以氘代氯仿为溶剂,测定氯甲基化聚砜CMPSF、改性聚砜PSF-AL和产物PSF-AA 3种聚合物的1H NMR谱,通过比较,进一步确认PSF-AA的化学结构;3)紫外分光光度法,测定功能化聚砜PSF-AA侧链上双齿席夫碱配基AA的键合量:取上清液样品,采用紫外分光光度法( λ=305 nm)测定反应液中未反应的AP浓度,从而计算出在聚砜侧链参与席夫碱反应的AP的量,此量即为功能化聚砜PSF-AA侧链上双齿席夫碱配基AA的键合量BA(Bonding amount,mmol/g)。 本研究在适宜的条件下所制得的PSF-AA,其AA的键合量为1.30 mmol/g。
在上述三步大分子反应中,聚砜的氯甲基化反应本课题组已多有研究,而醛基与伯氨基之间的席夫碱反应又易于进行,故为制得双齿席夫碱配基AA键合量高的功能化聚砜PSF-AA,本研究重点考察研究影响CMPSF与GA之间的酯化反应的主要因素。 固定其它反应条件,分别系列地改变反应溶剂种类和反应温度,考察这些因素对氯烷与羧基之间酯化反应(属亲核取代反应)的影响,优化反应条件,并探讨反应机理。
1.4.1 两种二元高分子-稀土配合物的制备 称取0.75 g Tb4O7,加入75 mL盐酸溶液(6 mol/L),加热搅拌使其溶解,静置冷却后加入5 mL H2O2,磁力搅拌2 h后,加热浓缩至有晶粒开始产生,冷却后,析出大量晶体,真空干燥至恒重,即得水合三氯化铽(TbCl3·6H2O)。 准确称取0.35 g功能大分子PSF-AA(AA的量为0.42 mmol),将其溶于50 mL DMAC中,再加入0.052 g三氯化铽晶体(0.14 mmol),也使之溶解;将该溶液体系置于60 ℃下,恒温搅拌反应10 h;加入沉淀剂乙醇,沉淀出产物,过滤,并用乙醇和蒸馏水交替洗涤后,真空干燥至恒重,即得二元高分子-稀土配合物PSF-(AA)3-Tb(Ⅲ)。
同样以Eu2O3为试剂,制得水合氯化铕(EuCl3·6H2O)[18],依上述步骤,制得二元高分子-稀土配合物PSF-(AA)3-Eu (Ⅲ)。
1.4.2 配合物荧光发射光谱的测定 分别配制配合物PSF-(AA)3-Tb(Ⅲ) 和PSF-(AA)3-Eu(Ⅲ)的DMAC溶液(使Tb3+离子及Eu3+离子的浓度均为4.0×10-5 mol/L);以Tb3+离子545 nm处的特征发射扫描PSF-(AA)3-Tb(Ⅲ)溶液的激发光谱,测得最佳激发峰在278 nm;然后使用荧光光谱仪,以最佳激发峰,测定该溶液的荧光发射光谱。 也以Eu3+离子620 nm处的特征发射扫描该溶液的激发光谱,测得最佳激发峰在312 nm,以此为最佳激发峰,测定PSF-(AA)3-Eu(Ⅲ)溶液的荧光发射光谱。
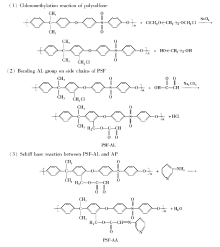
首先,以BCMB为氯甲基化试剂,使聚砜发生氯甲基化反应,制得氯甲基化聚砜CMPSF;然后以乙醛酸GA为试剂,使CMPSF的氯甲基与GA分子中的羟基发生酯化反应,获得侧链键合乙醛基团AL的改性聚砜PSF-AL;最后,以3-氨基吡啶AP为试剂,使改性聚砜PSF-AL侧链上的醛基与AP分子中的伯氨基发生席夫碱反应,从而在聚砜侧链上形成双齿席夫碱配基(该配基亚胺结构C=N中的N原子和吡啶N原子即为“双齿”,使得该配基可与稀土离子发生螯合配位),即获得了侧链键合有双齿席夫碱配基的功能化聚砜PSF-AA,上述制备功能化聚砜PSF-AA的化学反应过程如Scheme 1所示。
 | Scheme 1 Preparing PSF-AA |
{kind=link}
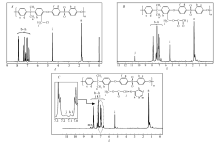
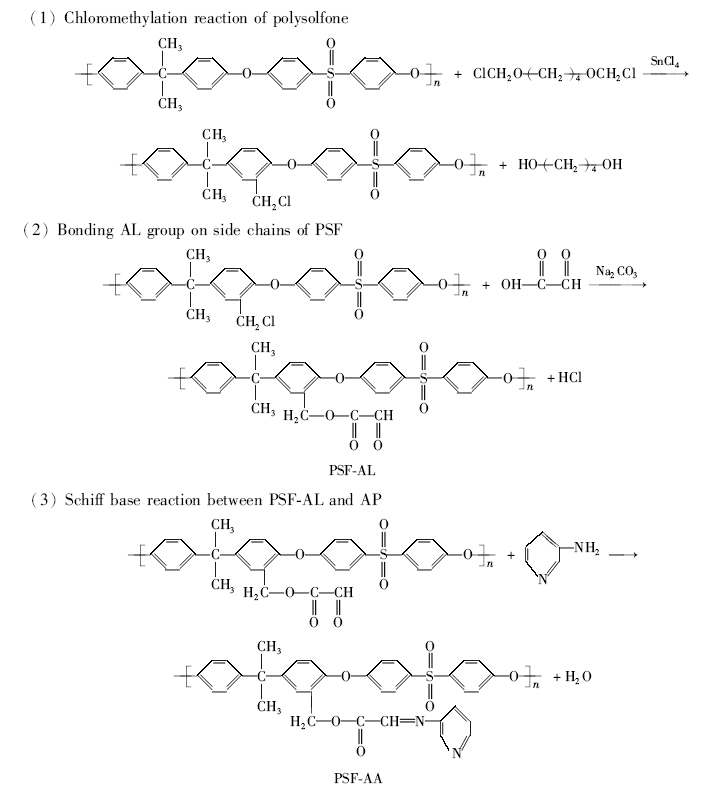
2.2.1 红外光谱图1给出了聚砜PSF、氯甲基化聚砜CMPSF、改性聚砜PSF-AL和功能化聚砜PSF-AA 4种聚合物的红外光谱。
 | 图1 4种聚合物的红外光谱Fig.1 FTIR spectra of four polymers a.PSF; b.CMPSF; c.PSF-AL; d.PSF-AA |
{kind=link}
在CMPSF的谱图中,除显示出聚砜所有特征吸收峰外,还于1442 cm-1处出现了氯甲基—CH2Cl中C—H键的面内弯曲振动吸收峰,于664 cm-1处出现了C—Cl键的伸缩振动吸收峰,于885 cm-1处还出现了苯环上在1、2、4号位上发生三元取代后的特征吸收峰[19,20],表明CMPSF的生成。
在改性聚砜PSF-AL的谱图中,吸收峰发生了明显变化。 1442和664 cm-1处的氯甲基的特征吸收峰明显减弱甚至消失;与此同时,在1730 cm-1处出现了酯基上的C=O键的伸缩振动吸收,在1675 cm-1处出现了醛基的特征吸收,2726 cm-1处则出现了醛基上C—H键的伸缩振动吸收峰,在1230 cm-1处出现酯基中C—O—C键的伸缩振动吸收峰。 上述谱峰数据的变化充分表明,CMPSF已与GA发生酯化反应,形成了侧链带有乙醛基的改性聚砜PSF-AL。
在功能化聚砜PSF-AA的谱图中,1675 cm-1与2726 cm-1处的醛基特征吸收已明显减弱甚至消失,而在1642 cm-1处出现了C=N键的伸缩振动吸收峰,充分证实大分子PSF-AL侧链上的醛基已与AP分子中的伯胺基发生了席夫碱反应,形成了侧链键合双齿席夫碱配基的功能化聚砜PSF-AA。
2.2.2 氢 谱 以氘代氯仿为溶剂,分别测定了氯甲基化聚砜CMPSF、改性聚砜PSF-AL与功能化聚砜PSF-AA的1H NMR谱,图2 A、2 B与图2 C分别给出了3种聚合物的1H NMR谱图,并标出了各吸收峰与3种聚合物结构中诸氢质子的对应关系。
 | 图2 3种聚合物的氢谱图Fig.2 1H NMR spectra of three kinds of polymers A.CMPSF; B.PSF-AL; C.PSF-AA |
{kind=link}
在CMPSF的谱图中,化学位移在 δ 6.831~7.883(b~h)范围内的一组峰,对应于PSF大分子主链苯环上的各种氢质子的特征共振信号;双酚A单体单元上甲基氢质子的化学位移 δ 1.734(a);氯甲基上氢质子(i)的化学位移 δ 4.541。
与CMPSF的谱图相比较,在改性聚砜PSF-AL的谱图中,除出现PSF的特征吸收峰外,在化学位移 δ 10.597处出现醛基—CHO氢质子的共振峰;伴随着氯甲基的酯化,原氯甲基上氢质子已转变为酯基结构中的甲基氢质子(i),其化学位移发生了变化,由 δ 4.541变化至 δ 5.333。 上述氢谱数据表明,经过CMPSF与GA之间的酯化反应,在聚砜侧链上已成功键合乙醛基团AL。
与改性聚砜PSF-AL的谱图相比,在PSF-AA的谱图中发生了如下变化:1) δ 10.597处醛基—CHO氢质子的共振峰已消失;2)在 δ 7.167(j)处出现了亚胺基—CH=N—氢质子的共振吸收峰;3)吡啶环上k与l处氢质子的共振吸收与聚砜主链苯环上各种氢质子的共振信号交叠;4)化学位移 δ 8.023和 δ 8.199属于吡啶环上另外两种氢质子m和n的特征共振吸收。 上述氢谱数据进一步证实,改性聚砜PSF-AL侧链的醛基已与AP分子中的伯氨基发生了席夫碱反应,形成了双齿席夫碱配基功能化的聚砜PSF-AA。
如前所述,在制备双齿席夫碱配基功能化的聚砜PSF-AA的三步大分子反应中,聚砜的氯甲基化是本课题组所建立的较为成熟的反应过程,而席夫碱反应又易于进行,故氯甲基化聚砜CMPSF与乙醛酸GA之间的酯化反应是制备功能化聚砜PSF-AA的关键步骤,本研究重点考察了主要条件对此反应的影响,以期优化反应条件,制备AA键合量高的功能化聚砜PSF-AA。
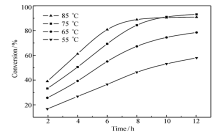
2.3.1 温度的影响 以DMF为溶剂,在不同温度下进行了CMPSF与GA之间的酯化反应,图3 给出了不同温度下CMPSF的氯甲基转化率随反应时间的变化曲线。
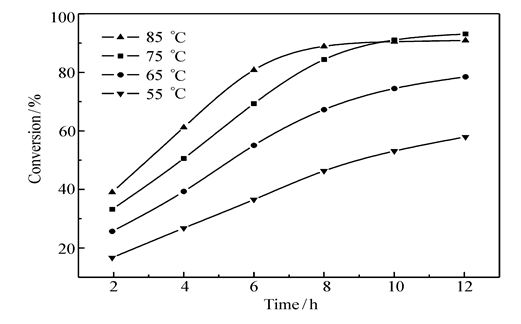 | 图3 不同温度下CMPSF的氯甲基转化率随反应时间的变化曲线Fig.3 Conversion of chloromethyl group on CMPSF at different temperaturesSolvent: DMF |
{kind=link}
从图3可以看出,在所选取的温度范围内,随着反应温度的升高,同一反应时间内氯甲基的转化率提高,符合反应动力学的规律,温度升高,反应速度加快。 但是,从图3还可发现另一规律:当温度低于75 ℃时,随反应时间延长,氯甲基的转化率在持续增高;当温度增至75 ℃时,反应12 h后,氯甲基的转化率增高的趋势变缓;当温度高于75 ℃时(如85 ℃),反应10 h后,氯甲基转化率趋于稳定不变的情形,致使10 h后氯甲基转化率反而低于75 ℃的氯甲基转化率。 对于一个放热反应,温度从动力学(正性)和热力学(负性)两个方面发挥影响,往往会出现上述规律,故推测CMPSF与GA之间的酯化反应可能为放热反应。 对上述实验事实做如下分析:1)在温度较低时,随着温度升高,反应速率加快,氯甲基转化率迅速提高,这是动力学因素所致。对于放热反应,升高温度会使平衡转化率降低,但在较低温度条件下,反应12 h,反应远未达到平衡,此时动力学因素占主导,故氯甲基转化率随反应时间仍呈现持续增高的趋势(反应12 h时,曲线仍陡峭);2)当温度升至75 ℃,反应12 h后,氯甲基的转化率增高的趋势变缓,意味着反应即将达到平衡;3)当温度高于75 ℃、升至85 ℃时,便显露出热力学的负面影响,或者说热力学因素变成了主导因素,10 h后反应基本达到了平衡,致使氯甲基转化率明显低于75 ℃的数值,曲线发生交错。 从图中4条曲线的走势看,85 ℃的平衡转化率应该是最低的。 综合上述动力学因素与热力学因素的影响,显然,适宜的反应温度应选择75 ℃。
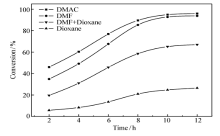
2.3.2 溶剂极性的影响 CMPSF的氯甲基与乙醛酸GA分子中羧基之间的酯化反应,属于氯烷的亲核取代反应,溶剂极性对氯烷的亲核取代反应具有较大的影响,故本研究选用了4种极性不同的溶剂DMAC、DMF、DMF+Dioxane(体积比1:1)、Dioxane实施了酯化反应。图4给出了在不同溶剂中进行反应时,CMPSF大分子上氯甲基的转化率随时间的变化曲线,表1 则列出了各种溶剂的介电常数与极性常数数据。
| 表1 各种溶剂的介电常数与极性常数 Table 1 Dielectric constant and polarity parameter for various solvents |
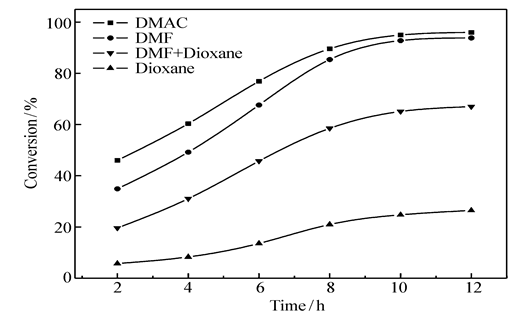 | 图4 使用不同溶剂时CMPSF氯甲基的转化率随时间的变化曲线Fig.4 Conversion chloromethyl group on CMPSF in different solventsTemperature:75 ℃ |
{kind=link}
图4显示,在相同的条件下,在4种溶剂中酯化反应速率快慢的顺序为:DMAC>DMF>DMF+Dioxane>Dioxane。 由表1数据可以看出,4种溶剂极性大小的顺序与上述亲核取代反应速率的顺序是一致的,即溶剂的极性越强,氯甲基的转化率越高,亲核取代反应速率越快。
CMPSF大分子链中氯甲基与乙醛酸GA分子中羧基之间亲核取代反应的实质是:在缚酸剂的存在下,氯甲基失氯变为碳正离子,与此同时,羧羟基失去氢质子,变为羧酸负离子亲核进攻基团,而后发生酯化反应,形成改性聚砜PSF-AL。 氯甲基失氯变为碳正离子的容易程度与溶剂的极性关系密切,溶剂的极性越强,溶剂与氯甲基之间的偶极-偶极相互作用越强,越有利于氯原子的脱去,越有利于亲核取代反应的进行,显然,溶剂的极性可促进亲核取代反应的进行,故在极性强的溶剂中,CMPSF与乙醛酸GA之间的酯化反应越容易进行。 本研究选择DMAC为适宜的反应溶剂,在75 ℃下反应12 h,可使CMPSF的氯甲基转化率达到96%,进一步经过席夫碱反应,制得了AA的键合量为1.30 mmol/g的功能化聚砜PSF-AA。
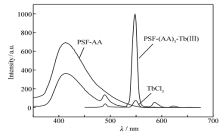
使功能化聚砜PSF-AA与Tb(Ⅲ)离子配位,得到二元配合物PSF-(AA)3-Tb(Ⅲ),将配合物溶于DMAC,配制成Tb(Ⅲ)离子浓度为4.0×10-5 mol/L的溶液,以最佳激发波长(见1.4.2节)测定其荧光发射光谱,同时也测定TbCl3溶液的荧光发射光谱,测定结果示于图5,图6则给出两溶液在紫外光辐射下光致发光的照片。
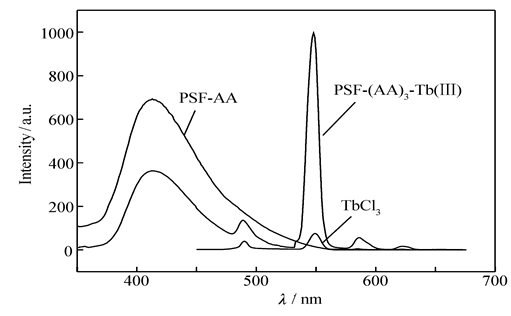 | 图5 二元配合物PSF-(AA)3-Tb(Ⅲ)、TbCl3和PSF-AA的荧光发射光谱Fig.5 Fluorescence emission spectra of binary complex PSF-(AA)3-Tb(Ⅲ), TbCl3 and PSF-AA, respectivelySolvent:DMAC; concentration of Tb(Ⅲ):4.0×10-5 mol/L |
{kind=link}
 | 图6 配合物PSF-(AA)3-Tb(Ⅲ)和TbCl3溶液光致发光的照片Fig.6 Photoluminescence photographs of PSF-(AA)3-Tb(Ⅲ) and TbCl3 solutions |
{kind=link}
图5显示,大分子配体PSF-AA本身在413 nm处有荧光发射。 从Scheme 1可以看到,在PSF-AA侧链双齿席夫碱配基AA的结构中,通过亚胺基C=N的纽带,左边酯羰基C=O与右边的吡啶环形成了一个较大的共轭体系,当紫外光照射时,PSF-AA会强烈地吸收光能,产生π-π*电子能级跃迁,电子激发至第二激发态,再通过振动弛豫过程降至第一激发态,然后发射出自身的荧光。 但是,当大分子配体PSF-AA与Tb(Ⅲ)离子配位形成二元配合物PSF-(AA)3-Tb(Ⅲ)后,其本身的荧光发射大为减弱,这是发生了分子内能量转移的显著信号,与有关文献报道的情况类似[21,22]。
从图5与图6可以看出: 1)二元配合物PSF-(AA)3-Tb(Ⅲ)的荧光发射谱带的位置和形状与Tb(Ⅲ)离子相同,表明配合物发射出Tb(Ⅲ)离子的特征荧光,谱图中主要显示出Tb(Ⅲ)离子490 nm处的5 D4→7 F6跃迁、549 nm处的5 D4→7 F5跃迁、585 nm处的5 D4→7 F4及623 nm处的5 D4→7 F3跃迁所导致的发射,其中5 D4→7 F5跃迁所产生的发射最强; 2)二元配合物的荧光强度远比TbCl3强,提高约14倍; 3)二元配合物PSF-(AA)3-Tb(Ⅲ)发生了强烈的光致发光现象,在紫外光辐射下,明显发射出Tb(Ⅲ)离子的绿色荧光;而TbCl3荧光发射很弱,溶液几乎是无色的。 上述实验事实充分表明,大分子配体PSF-AA对Tb(Ⅲ)离子的荧光发射产生了强烈的敏化作用,即产生了显著的 Antenna 效应,表明大分子配体侧链上的双齿席夫碱配基AA的三线态能级与Tb(Ⅲ)离子的共振能级是相互匹配的[23,24]。 PSF-AA大分子配体(实体是配基AA)吸收能量后,处于最低激发单重态( S1)的电子大部分经系间窜越至三重态,再由最低激发三重态( T1)以非辐射方式将能量传递给Tb(Ⅲ)离子,然后受激的Tb(Ⅲ)离子再以辐射方式跃迁到基态能级,从而发射出Tb(Ⅲ)离子的特征荧光(绿光)。 与此同时,随着上述发生分子内的能量转移,大分子配体PSF-AA自身的荧光发射则大为减弱。
作为对比,本研究也制备了PSF-AA与Eu(Ⅲ)离子的二元配合物PSF-(AA)3-Eu(Ⅲ),也测定了该配合物及EuCl3溶液的荧光发射光谱,示于图7。
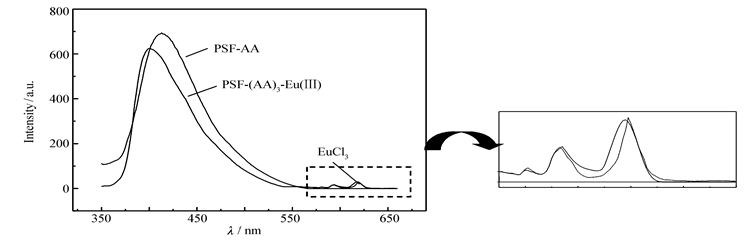 | 图7 二元配合物PSF-(AA)3-Eu(Ⅲ)、EuCl3和PSF-AA的荧光发射光谱Fig.7 Fluorescence emission spectra of binary complex PSF-(AA)3-Eu(Ⅲ), EuCl3 and PSF-AA, respectivelySolvent: DMAC; concentration of Eu(Ⅲ):4.0×10-5 mol/L |
{kind=link}
从图7可以看出:1)在二元配合物PSF-(AA)3-Eu(Ⅲ)的荧光发射光谱中,大分子配体PSF-AA的荧光发射依然强烈,与未配位时的情形相差不多,意味着配合物分子内几乎没有发生能量转移;2)二元配合物PSF-(AA)3-Eu(Ⅲ)在580~620 nm范围内的出峰情况(峰的位置与强度)几乎与EuCl3溶液相差无几,没有红光产生,说明大分子配体对Eu(Ⅲ)离子的荧光发射没有敏化作用,显示出大分子配体侧链上的双齿席夫碱配基AA其三线态能级与Eu(Ⅲ)离子的共振能级是严重不匹配的。
总之,初步的探索研究表明,本研究所制备的双齿席夫碱配基功能化聚砜PSF-AA对稀土离子具有双重功能,即既具有配位螯合作用,又对其荧光发射具有显著的敏化作用。 但是,与前文所制备的功能化聚砜PSF-SB相比[18],由于双齿席夫碱配基的结构不同,AA强烈敏化Tb(Ⅲ)离子的荧光发射,对Eu(Ⅲ)离子的荧光发射则不产生敏化作用,二元配合物PSF-(AA)3-Tb(Ⅲ)发射绿光,而配合物PSF-(AA)3-Eu(Ⅲ)不发射红光。 关于上述高分子-稀土配合物详细的光物理过程与机理、结构与性能关系将另文报道。
本研究通过分子设计,经过三步大分子反应(氯甲基化反应、酯化反应和席夫碱反应),在聚砜侧链合成了具有一定结构的席夫碱配基AA,成功地制备了双齿席夫碱配基功能化的聚砜PSF-AA。 氯甲基化聚砜与乙醛酸之间的酯化反应属于氯烷的亲核取代反应,适宜的溶剂为极性强的 N, N-二甲基乙酰胺,适宜的反应温度为75 ℃。 双齿席夫碱配基AA对Tb(Ⅲ)离子具有配位螯合与荧光敏化双重功能。 双齿席夫碱配基AA自身具有较强的荧光发射,但是当PSF-AA与Tb(Ⅲ)离子配位生成配合物后,可有效地发生光分子内能量转移,配基AA可强烈地敏化Tb(Ⅲ)离子的荧光发射,故二元高分子-稀土配合物PSF-(AA)3- Tb(Ⅲ)是发射绿光的光致发光材料。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|