
XU Bo, SUN Yanjie, DU Feipu, et al. Synthesis of ansa-(Cyclopentadienyl)(adamantylamido) titanium Complex for Propylene Polymerization[J]. Chinese Journal of Applied Chemistry, 32(9): 1028-1032


合成了新型限制几何构型四甲基环戊二烯金刚烷氨基二甲基钛配合物(1-Adamantyl)NSiMe2(C5Me4)]TiMe2(c)。 通过1H NMR、元素分析以及单晶衍射分析等技术手段确定了该配合物的结构。 与其叔丁基氨配合物 tBuNSiMe2(C5Me4)]TiMe2(a)对比,在修饰甲基铝氧烷(MMAO)/2,6-二叔丁基-4-甲基苯酚(BHT)活化下进行丙烯聚合,探讨了氨基配体结构及聚合条件对聚合性能的影响。 在保持准活性聚合的条件下,金刚烷胺基配合物c的催化活性为562 kg(PP)/(mol(Ti)·h),为叔丁胺基配合物a的2倍以上,说明金刚烷胺的引入有助于提高体系的链增长速率。
(1-Adamantyl)NSiMe2(C5Me4)]TiMe2(c) was synthesized and characterized by elemental analysis,1H NMR, and single-crystal X-ray analysis. Propylene polymerization was conducted with complex c using modified methyaluminoxane(MMAO)/butylated hydroxytoluene(BHT) as a cocatalyst to compare the polymerization activity of complex c with that of tert-butylamido complex( tBuNSiMe2(C5Me4)]TiMe2)(a). The c-heptane system shows an activity of 562 kg(PP)/(mol(Ti)·h) which is two times higher than that of a-heptane system indicating that the activity is improved by the introduction of 1-adamantyl substituent to the amido ligand.
1990年Bercaw和Okuda[1,2]首次报道了一种半茂金属催化剂,N原子通过桥与环戊二烯相连形成了配体的骨架结构,被称为“柄形单环戊二烯胺配合物”(ansa-monocyclopentadienylamido complex CpA)。 CpA配合物可以以很高的活性催化合成线形聚乙烯,而且具有优异的共聚能力,可以催化乙烯、 α烯烃、降冰片烯以及苯乙烯等进行共聚合反应[3,4,5],这类配合物具有的另一个主要特征是:配位空间只能朝一个方向打开,因此也被称为限制几何构型催化剂(constrained geometry catalyst CGC)。
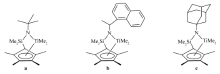
早期合成的CGC 配合物 tBuNSiMe2(C5Me4)]TiMe2(a)(见Scheme 1)可以催化丙烯聚合,一般只会得到无规聚丙烯。 而后大量的研究工作集中于通过控制聚合条件、改变催化剂的结构来获得CGC对丙烯聚合的活性以及立构选择性[6]。
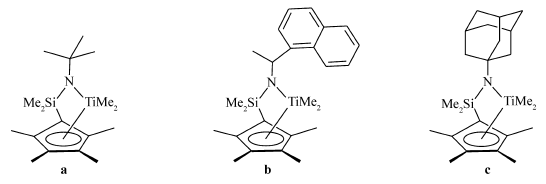 | Scheme 1 Constrained geometry catalysts |
{kind=link}
蔡等[7]曾报道过C1对称的[NCHMe(Naphthyl)SiMe2(C5Me4)]TiMe2(b),在以dMMAO为助催化剂的体系中催化丙烯聚合,利用胺上的不对称位阻效应使增长链发生回跳,可以获得[mm]=0.61的富含等规组分的聚丙烯。 并且反应是以活性聚合的性质进行的,但是胺基上的吸电子取代基明显降低了催化剂的催化活性。 Canich等[8]报道过携带金刚烷氨基的CGC型二氯钛配合物(1-Adamantyl)NSiMe2(C5Me4)]TiCl2,可以获得相对分子质量达到5.0×106的无规聚丙烯。 基于此,我们在N原子上引入具有良好对称性的金刚烷基代替叔丁基,合成CGC型二甲基钛配合物,确定该配合物的结构,以此研究氨基取代基部分的位阻和电子效应对二甲基钛配合物催化性能的影响。
所有反应均使用标准Schlenk技术,在高纯N2气保护下进行。
1,2,3,4-四甲基-1,3-环戊二烯(1,2,3,4-tetramethyl-1,3-cyclopentadiene)85%以及1-金刚烷胺(1-Amantadine)95%购自武汉长成试剂有限公司;二氯二甲基硅烷(SiMe2Cl2)、正丁基锂( n-BuLi)、甲基锂(MeLi)、四氯化钛(TiCl4)、甲基溴化镁(MeMgBr)均为化学纯,皆购自上海百灵威试剂有限公司。
Bruker AVANCE-400型核磁共振波谱仪(德国Bruker公司),400 MHz;Rigaku RAXIS Ⅳ型X射线晶面探测仪(美国Rigaku公司),K α为Mo靶的波长( λ=0.071059 nm);PL-GPC 220型高温凝胶渗透色谱仪(Gel Permeation Chromatography,英国Agilent Technologies公司),150 ℃,溶剂为三氯苯(1,2,3-Trichlorobenzene)。
1.2.1 (C5Me4)SiMe2Cl的合成 按照文献[8]方法合成,取四甲基环戊二烯5 g起始反应,得到淡黄色油状的(C5Me4)SiMe2Cl:8.07 g,产率为91.9%。1H NMR(CDCl3), δ:7.28;3.12(s,1H,Cp);2.03(s,6H,Cp-CH3),1.86(s,6H,Cp- CH3),0.28 (s,6H,SiCH3)。
1.2.2 (1-Adamantyl)NHSiMe2(C5Me4)的合成 取2.32 g(15.0 mmol)1-金刚烷胺固体,加入适量乙醚搅拌分散,0 ℃下逐滴加入6.8mL n-BuLi(16.5 mmol,2.4 mol/L),室温下持续反应3 h,得到白色金刚烷胺锂盐悬浮液。 另取3.22 g(15.0 mmol)(C5Me4)SiMe2Cl溶于适量乙醚中,0 ℃下将上面得到的悬浮液转移至烧瓶中,室温反应过夜。 除去溶剂后用正己烷萃取,旋蒸出溶剂,得到黄色油状物(1-Adamantyl)NHSiMe2(C5Me4):4.27 g,产率为86.7%。1H NMR (CDCl3), δ:7.28;2.07(s,3H,Ad);2.00(s,6H,Ad);1.98(s,6H,Ad); 2.99(s,1H,Cp);1.84(s,6H,Cp-CH3);1.60(s,6H,Cp-CH3);1.13 (s,1H,NH);0.05(s,6H,SiCH3)。
1.2.3 [ N (1-Adamantyl)SiMe2(C5Me4)]TiMe2的合成 取1.32 g (1-Adamantyl)NHSiMe2(C5Me4)(4.0 mmol)溶于适量乙醚中,0 ℃下缓慢滴加11.2 mL的MeLi(18 mmol,1.6 mol/L),室温反应3 h,得到黄色锂盐浊液。 另取0.45 mL的TiCl4(4.0 mmol,189.68 g/mol,1.73 g/mL)溶于少量正己烷中,而后将上步所得黄色悬浊液转移至TiCl4溶液中。 反应30 min后,除去溶剂,用正己烷萃取,取上层深棕色浊液,滴加2.67 mL的MeBrMg(8 mmol,3.0 mol/L),常温反应3 h后,除去溶剂后用正己烷萃取,取上层黄色清液,浓缩后置于-30 ℃下重结晶,获得黄色晶体[( N (1-Adamantyl)SiMe2(C5Me4))TiMe2(c):0.54 g(1.3 mmol),产率为32.2%。1H NMR (CDCl3,CDCl3), δ:7.28; 2.17(m,9H,Cp-CH3,Ad);2.12(s,6H,Ad);1.92(s,6H,Cp-CH3);1.73(s,6H,Ad);0.49(s,6H, SiCH3);0.18(s,6H,TiCH3)。 C23H39NSiTi元素分析实测值(计算值)/%:C 68.75(68.12),H 9.88(9.69),N 2.87(3.45)。
助催化剂中的烷基铝会造成链转移反应而使体系丧失活性聚合特性。 Shiono等[9]曾使用去除烷基铝的改性甲基铝氧烷(dMMAO)作为该催化体系的助催化剂,实现了丙烯的活性聚合。 但制备dMMAO的过程繁琐,使用MMAO/BHT作为本实验的助催化体系,在反应前将MMAO溶液和BHT以一定比例混合以除去其中的烷基铝组分。 由于BHT羟基邻位的位阻效应,过量的BHT以及反应生成的烷基铝苯氧化合物并不会对聚合反应造成影响[10]。 以此获得类似dMMAO的助催化体系实现丙烯的活性聚合。
本实验采用半间歇法[9]进行丙烯聚合,即在Schlenk瓶中加入规定量的溶剂、MMAO/BHT溶液,再以恒定压力通入丙烯直至其达到饱和,随即加入催化剂溶液,聚合反应开始进行,保持丙烯压力恒定,通过流量仪记录其瞬时速率与累计流量。 当达到规定的时间或者产生一定聚合物量时,立即用酸性乙醇淬灭。 得到的聚合物经过充分洗涤以除去聚合物中残留的催化剂和助催化剂组分,将其在70 ℃环境下进行真空干燥8 h以上,称重后即可计算出催化剂的活性(kg(PP)/(mol(Ti)·h))。
配合物c氢谱数据见其合成部分。 其中金刚烷胺中的次甲基质子峰和四甲基环戊二烯甲基质子峰重叠,造成 δ 2.12处的峰变宽,而其积分面积符合相应质子数目的叠加。 另外,可以观察到图中钛甲基和硅甲基分别在0.18和0.49处出现一个单峰,说明配合物在溶液中呈镜面(Cs)对称,相应的茂甲基质子峰以及金刚烷质子峰也印证了该结论。 金刚烷中3种质子出峰清晰,且每种质子处于相同的化学环境中,可以说明金刚烷部分绕N—C轴旋转顺利,金刚烷的引入并未改变配合物的对称结构。
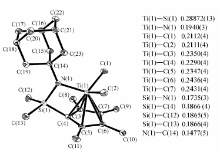
配合物c的单晶是在正己烷溶液中-30 ℃下放置7 d析出的,晶体呈黄色透明块状,分子结构见图1,单晶数据见表1。
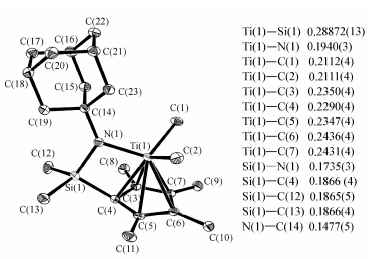 | 图1 配合物c的分子结构图Fig.1 The molecular structure of complex c |
{kind=link}
配合物c的晶体结构显示,其在固体状态下钛原子分别和同一分子的茂环和氮原子配位,不存在分子间配位。 键角方面,四边形Ti(1)—N(1)—Si(1)—C(4)内角和为359.79°;以N(1)为顶点的及其与C(14)、Si(1)、Ti(1)连线为边的三角之和为359.86;以上数据表明,N(1)、Ti(1)、Si(1)、C(4)、C(14)5个原子位于同一面内。 相似地,环戊二烯五元环内角和为539.98°,也可以证明其五碳共面,且上述两个面相互垂直。 上述钛氮硅原子所在平面即为配合物的对映面。键长方面,Ti(1)与C(4)的距离最近,与C(5)、C(6)距离依次变远。由于硅桥的作用,使钛原子所处位置偏离了茂环中心垂线,配位空间只能向图示钛甲基方向打开。 另外,钛原子以 η5方式和茂环配位,说明金刚烷胺基的引入并未改变限制几何构型催化剂的基本骨架结构。
| 表1 配合物c的晶体结构数据 Table 1 Crystallographic data and parameters for complex c |
丙烯聚合以配合物a作为参比。 按照文献[8]方法合成。
对比以上聚合结果可以发现随着温度的升高,两种体系聚合活性均明显提高(见表2)。 且配合物在甲苯溶液中比同等条件的庚烷溶液中有着更高的催化活性。 对于配合物c,在25 ℃下得到的聚合物的相对分子质量分布窄,小于1.5。温度升高至50 ℃时,两种体系均出现了明显的链转移,相对分子质量分布变宽,分子链数目超过了催化剂分子数。 50 ℃下配合物c的催化活性和活性链数明显高于a,可能的原因是,其聚合反应速率过高,聚合热无法及时通过油浴释放,体系温度上升,而温度的增加又继续使聚合速率增加,最终导致了反应初期的“加速效应”,使体系实际温度偏离预设温度。 当铝与钛摩尔比从200升为400时,聚合活性增加,加速现象明显,放热剧烈,导致生成的聚合物相对分子质量低,且计算所得聚合物链数目很高。
| 表2 不同条件下配合物a、c引发丙烯聚合结果 a Table 2 Results of propylene polymerization with complex a and b under different conditions a |
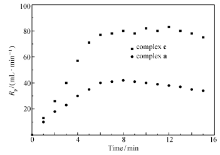
反应初期的“加速效应”会使反应变得不可控。 因此我们在其较低聚合速率下考察其动力学特征。图2为配合物a和c在正庚烷中25 ℃(表2,entry 1 and entry 5)的时间-聚合速率曲线。 图中可以观察到2个曲线均呈上升-平稳的趋势,6 min后丙烯通入速率保持恒定,获得的聚合物相对分子质量分布都较窄,高分子链数近似于催化剂投入量,说明该条件下2个体系未出现明显的链转移,25 ℃时仍具有明显的准活性聚合特征。 配合物c的丙烯消耗速率约为配合物a的两倍,且产生的聚合物的质量以及相对分子质量都近似为2倍关系,据此可以说明相比于叔丁基的CGC,携带金刚烷基有助于提高链增长速率而使其催化活性增加。
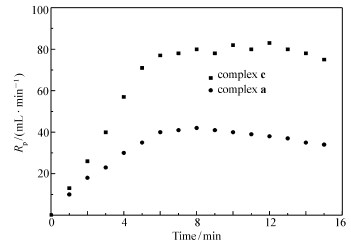 | 图2 丙烯消耗速率-时间曲线Fig.2 Rate-time profiles of propylene polymerization with complexs a or c |
{kind=link}
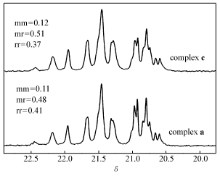
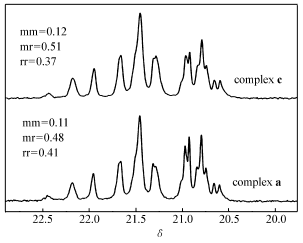 | 图3 聚丙烯甲基部分碳谱图Fig.3 13C NMR spectra of methyl region of PP |
{kind=link}
图3为配合物a、c分别在正庚烷中,25 ℃下催化合成的聚丙烯13C NMR谱。 两种配合物均只得到了无规聚丙烯。 而对配合物c,间规组分(rr)比例减少。 可能是由于大位阻基团1-金刚烷的引入,造成配合物茂环和氨基部分位阻差异降低,降低了配位点的面对称性,进而导致配合物的间规选择性降低。
合成了(1-Adamantyl)SiMe2(C5Me4)]TiMe2配合物c,通过了1H NMR和EA的鉴定,并通过X射线单晶衍射分析确定了其空间配位结构。 结果表明,配合物c在溶液中呈CS对称,钛原子与茂环呈 η5配位方式。 在MMAO/BHT的激活下,配合物c可以催化丙烯进行准活性聚合生成无规聚丙烯。 在同等聚合条件下配合物c的催化活性和耐链转移特明显高于其叔丁氨基类似物a,而生成聚丙烯中的间规组分[rr]相比于配合物a有所下降。 研究表明,CGC配合物氨基取代基的电子和位阻效应对配合物的催化行为有着显著的影响,为设计合成高活性CGC催化剂和超高分子量聚合物建立了研究工作基础。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|