

WEI Zanbin, WANG Jinchi, JIANG Xia, et al. Experimental and DFT Studies of Pyridine-4-carboxylic Acid (2,4-dihydroxy-phenylethylidene)-hydrazide Schiff Base:Synthesis, Crystal Structure, Properties and Quantum Chemistry Calculation[J]. Chinese Journal of Applied Chemistry, 32(9): 1014-1021


合成了一种酰腙类Schiff碱2,4-二羟基苯乙酮缩异烟酰腙(C14H13N3O3,H2L),经元素分析、红外光谱、紫外光谱、荧光光谱和热重分析等技术手段进行了表征。 用X射线单晶衍射测定了它的晶体结构,该晶体属单斜晶系,C2/c空间群,晶胞参数 a=2.0102(2) nm, b=0.75891(8) nm, c=1.9530(2) nm, α= 90°, β=111.481(12)°, γ=90°, V=2.7725(5) nm3, Z=4, Dc=1.4292 g/cm3, R1=0.0422, wR2=0.1113, F(000)=1256。 同时进行了量子化学计算研究。 使用Gaussian09量子化学程序包, 在密度泛函理论(DFT)的B3LYP/6-31G(d)水平,对化合物的分子结构进行全参数优化计算,获得了热力学参数和几何结构参数,对分子的总能量及前线分子轨道、Mulliken电荷分布进行了分析讨论;同时,用TD-DEF方法计算了化合物的电子吸收光谱和荧光发射光谱。
A hydrazone-type Schiff base pyridine-4-carboxylic acid (2,4-dihydroxy-phenylethylidene)-hydrazide(H2L) was synthesized and characterized by elemental analysis, IR, UV, FL spectra, TGA and X-ray diffraction single crystal analysis which result showed that the crystal of HL·1.5H2O belongs to the monoclinic system, space group C2/c with cell parameters a=2.0102(2) nm, b=0.75891(8) nm, c=1.9530(2) nm, β=111.481(12)°, V=2.7725(5) nm3, Z=4, Dc=1.4292 g/cm3, R1=0.0422, wR2=0.1113, F(000)=1256. The compound formed 3-D supermolecule via intermolecular hydrogen bondings. The quantum chemical calculation was performed by means of Gaussian 09 program at B3LYP/6-31G(d) basis set.After optimization of molecular geometry, some important thermodynamic parameters and structural parameters were obtained. The molecular dipole moment, Wiberg bond order, Mulliken charge population and frontier molecular orbital energy have been analyzed and discussed. TD-DFT method is used to calculate the absorption and fluorescence spectra of the compound. CCDC 994067.

长期以来,Schiff碱由于容易合成、具有丰富的结构和广泛的应用前景而备受研究者关注,新型Schiff碱及其配合物层出不穷[1,2,3,4,5,6,7,8,9,10]。 酰腙类化合物是由酰肼与醛或酮缩合而成的一类Schiff碱类化合物,以N、O原子与金属离子配位与生物环境相似,因而表现出良好的生物活性;有较强的配位能力和多样的配位方式;由于次胺基氮原子的孤对电子与酰基和亚胺基双键形成了p-π共轭体系所以比普通的Schiff碱化合物更稳定;某些酰腙类化合物的金属配合物具有良好的发光和催化活性,在材料科学领域有着潜在的应用价值。 2,4-二羟基苯甲醛缩异烟酰腙Schiff碱(H2L)(Scheme 1)是一种多齿配体,VaramY等[11]曾经研究了它与稀土离子的配位作用,但H2L的晶体结构未见报道。 本文培养了2,4-二羟基苯甲醛缩异烟酰腙单晶,经X射线单晶衍射仪测定了它的晶体结构,分析了其光谱性质和热稳定性。 目前,密度泛函理论(DFT)已发展成为一种重要的量子化学计算方法,它能提供非常准确的电子分布和分子几何构型等信息,对于研究化合物分子的构效关系具有重要的意义[12,13,14,15,16]。 因此,本文根据晶体数据,采用密度泛函理论优化了化合物的几何结构并计算了重要的键级、分子轨道和电荷布局等参数以及电子吸收光谱和荧光发射光谱,以便为其进一步的应用提供理论参考。
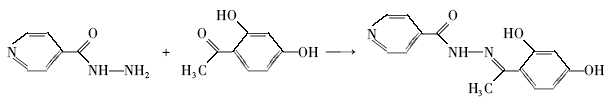 | Scheme 1 Reaction scheme for the H2L |
Vario EL型元素分析仪(德国Elementar公司);STA 409 PC型综合热分析仪(德国Netzsch公司); is10型FT-IR红外光谱仪(美国Nicolet公司);UV-1800PC型紫外-可见分光光度计(上海美谱达公司);CARY/Eclipse型荧光分光光度计(美国Varian公司);Xcalibur Sapphire3 Gemini ultra型单晶衍射仪(英国Agilent公司)。 所用试剂均为分析纯。
化合物H2L·1.5H2O的合成参考文献[11]。 将1.3714 g(10 mmol)异烟酰肼溶于20 mL热的无水乙醇中,滴入10 mL含1.5215 g(10 mmol)2,4-二羟基苯乙酮的乙醇溶液,加入0.5 mL冰醋酸,65 ℃下加热回流3 h,冷却,放置1 d后,析出大量浅黄色片状晶体,抽滤,并用无水乙醇洗涤,然后置于干燥器中用浓硫酸干燥备用。 产率约63%。 C14H13N3O3·1.5H2O元素分析实测值(计算值)/%:C 56.33(56.37),H 5.46(5.41),N 14.02(14.09)。 红外光谱数据(KBr), σ/cm-1:3520,3430,3276,1670,1617,1578,1507,1472,1327,1270,842,677。
取0.28 mm×0.20 mm×0.10 mm的化合物H2L·1.5H2O的单晶置于Agilent Xcalibur Sapphire3 Gemini ultra单晶衍射仪样品架上,以石墨单色器单色化的Mo Kα射线( λ=0.071073 nm),极限指数为-21≤ h≤23,-9≤ k≤6,-23≤ l≤21,在293(292) K温度下,以 ω扫描方式在3.41°< θ<25.00°范围内收集到5558个衍射点,其中2430个独立衍射点( Rint=0.0172),2136个I>2 σ( I)的可观察点。 晶体结构采用SHELXS-97程序由直接法解出,并采用SHELXL-97程序[17]通过全矩阵最小二乘法对非氢原子坐标和各向异性热参数进行精修,以理论加氢法确定氢原子的位置,晶体学数据见表1,部分键长和键角列于表2和表3。 CCDC 994067
| 表1 H2L·1.5H2O的晶体学数据及精修参数表 Table 1 Crystal data and structure refinement parameters for compound H2L·1.5H2O |

根据实测化合物的晶体结构,选取晶体结构中的1个分子作为初始模型,运用Gaussian09 量子化学程序包[18],采用密度泛函理论(DFT) B3LYP方法,在6-31G(d)水平上对分子进行全优化计算。 计算采用的原子坐标来自晶体结构数据,计算中收敛精度取程序默认值。 计算结果表明,所得的优化几何构型均对应于势能面上的能量极小点(振动无虚频)。
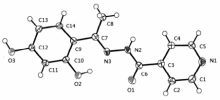
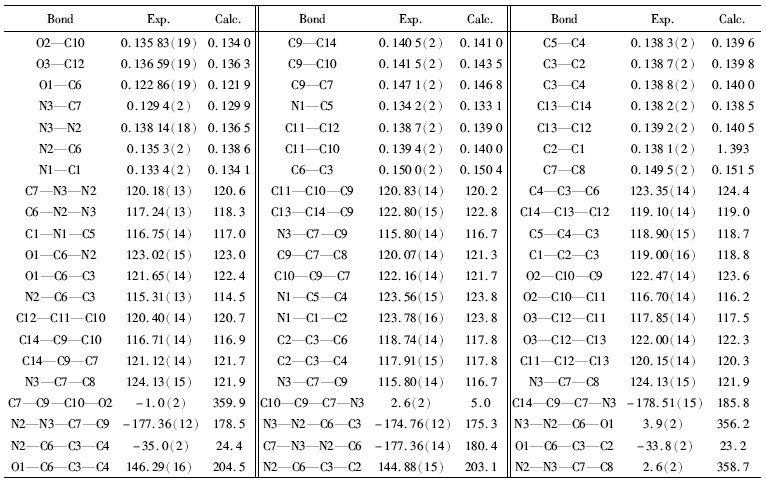
晶体结构分析表明,标题化合物晶体属单斜晶系,C 2/c空间群,H2L的分子结构如图1所示。 O(1)—C(6)键长为0.12286(19) nm,属典型C=O双键,说明该酰腙化合物为酮式结构;但是O1—C6—C3—C2扭转角为-33.77°,说明C=O与吡啶环未发生共轭作用,所以C3—C6仍保持典型单键特征,键长0.1500(2) nm,与乙基的C7—C8(0.149 5(2) nm)键长相近,而C7—C9键长0.1471(2) nm,比前二者则小得多,这是因为O2与N3之间的分子内氢键(0.25411 nm)导致亚胺基C=N与苯环共平面而存在较强的共轭作用。 N3、C7、C9、C10和C14基本共平面,扭转角N3—C7—C8—C10为2.52°、N3—C7—C8—C14为178.51°。 N3—C7键长0.1294(2) nm,属典型C=N双键;次胺基与羰基和亚胺基存在p-π共轭,C6—N2—N3键角为117.24(13)°扭转角O1—C6—N2—N3为3.89°,N2—N3—C7—C9为 -177.34°,导致N2—C6键长(0.1353(2) nm)与吡啶环内C—N键长(0.1334(2)~0.1342(2) nm)相近。 O2—C10和O3—C12键长分别为0.13583(19)和0.13659(19) nm,介于C—O单键和C=O双键之间,说明羟基氧与苯环存在共轭作用。
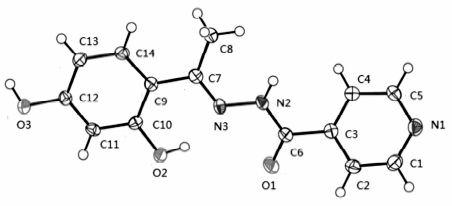 | 图1 H2L的分子结构(椭球率30%)Fig.1 Molecular structure of H2L with 30% probability displacement |
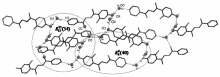
晶体中存在丰富的分子间氢键(图2)。 晶格水O4与羟基O3之间(0.2841 nm)、羟基氧O3与晶格水O5之间(0.2614 nm)和晶格水O5与另一分子的吡啶环N1原子之间(0.2806 nm)的氢键构成
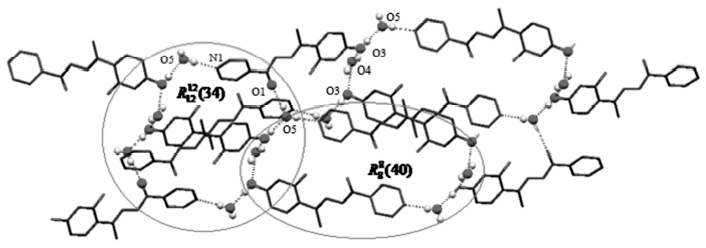 | 图2 H2L·1.5H2O晶体中的氢键Fig.2 Hydrogen bonds in the crystal of H2L·1.5H2O |
2.2.1 结构优化、热力学参数和CN键级 优化结构的部分几何参数列于表2,计算值与实验值比较接近,表明计算方法是可靠的。C=N的Wiberg 键级(BOC=N):C7—N3:1.652;C5—N1:1.430;C1—N1:1.417。 C7—N3的键级大于1.500,属典型C=N双键,该计算值和实验值一致。 C5—N1和C1—N1键级小于1.500,是因为吡啶环内原子共轭的结果。 分子的最高占据轨道能量 EHOMO=-5.660 eV,最低空轨道能量 ELUMO=-1.878 eV。 标准焓 Hθ=-25332.488 eV,标准自由能 Gθ=-25334.291 eV,恒容热熔
| 表2 标题化合物的主要键长(nm)、键角(°)和二面角(°) Table 2 Selected bond lengths(nm), angles(°) and torsion angle(°) of title compound |
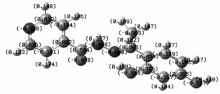
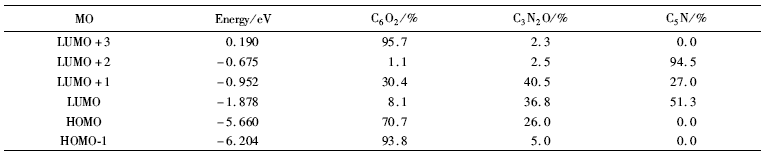
2.2.2 前线分子轨道及组成 标题化合物前线分子轨道如图3所示。 最高占据轨道能量 EHOMO=-5.660 eV,最低空轨道能量 ELUMO=-1.878 eV,Δ E=ELOMO- EHUMO=3.782 eV,差值较小,说明化合物具有较高的活性。 将分子分为3部分,分别为二羟基苯C6O2、酰腙基C3N2O和吡啶基C5N,运用自然键轨道处理方法得到了分子轨道中各个碎片原子所占的比例(表3)。 最高占据轨道HOMO电子云主要集中在二羟基苯(70.7%)和酰腙基团(26.0%),根据分子轨道理论, 最高及其附近的占有分子轨道具有优先提供电子的作用[19],说明此部分优先提供电子与金属离子形成配位键,也是发生氧化过程的中心。 LUMO的主要成份几乎遍布于除氢原子外的整个化合物,说明这些原子的空轨道可作为电子的受体,成为化合物反应的活性中心。 对LUMO的贡献二羟基苯降至8.1%,而吡啶环增至51.3%,说明电子由HOMO向LOMO跃迁时,主要发生的是电子由二羟基苯向吡啶环的转移。
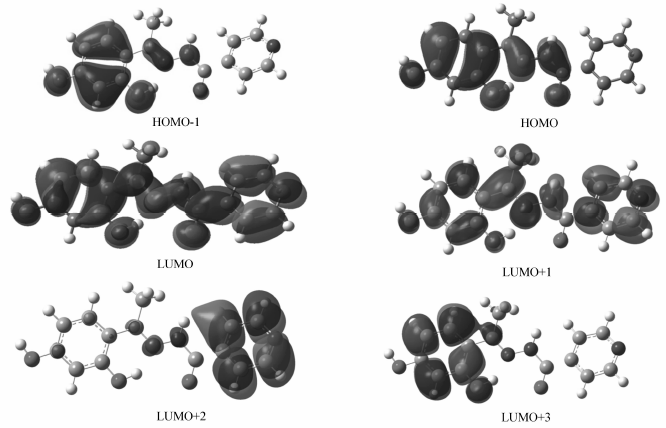 | 图3 标题化合物的前线分子轨道Fig.3 The schematic diagram of frontier MO for title compound |
| 表3 化合物的前线分子轨道能量(eV)和分子碎片对该分子轨道贡献(%) Table 3 Calculated energy levels(eV) of some frontier MO and contribution of molecular fragments(%) |
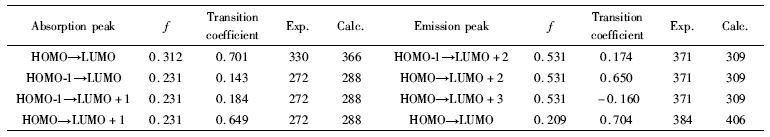
| 表4 标题化合物的吸收峰(nm)和发射峰(nm) Table 4 Absorption and emission peaks(nm) of title compound |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
2.2.3 Mulliken电荷分布 化合物的电荷分布见图4。 高负电荷密度区域往往就是分子的活性区域。 分子中所有杂原子都带有较高的负电荷,Mulliken净电荷之和(Σ Q( X))为-3.057e,其中O1、O2和O3分别为-0.478e、-0.646e和-0.637e;N1、N2和N3分别为-0.398e、-0.498e和-0.400e。 说明这6个位点是亲核反应中心,可与金属离子的空轨道形成配位键或与生物大分子作用。 N1尽管带有负电荷,然而它对HOMO的贡献非常小,所以与金属离子的配位能力较弱。 而羟基和酰腙基团对HOMO的贡献很大,所以它们构成的负电荷空腔将成为金属离子优先进攻的位点而形成配位键。 而高负电性的羟基氧(O2和O3)将成为抗氧化性的活性中心并能与生物分子形成氢键而具有一定的生物活性。
 | 图4 化合物的Mulliken电荷分布Fig.4 The Mulliken charge population of title compound |
{kind=link}
化合物的IR光谱中,3520 cm-1处尖锐的吸收峰是羟基的伸缩振动,3430 cm-1属结晶水的伸缩振动,3276 cm-1吸收峰则由酰腙片段的 νNH产生。 1670和1617 cm-1强吸收峰分别归属酰胺基的 νC=O和亚胺基的 νC=N,1327和1270 cm-1可指认为 νC—N和酚羟基 νAr—O。
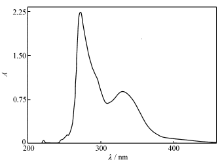
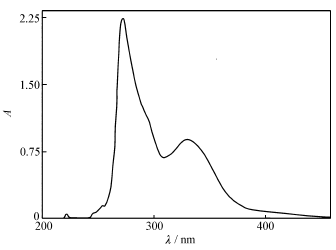 | 图5 化合物的紫外光谱图Fig.5 UV spectrum of title compound |
{kind=link}
在结构最优化数据基础上选择含时密度泛函的方法进行能量再次优化,计算了电子吸收光谱和荧光发射光谱各跃迁的理论值和跃迁系数 ,与实验值一同列入表4。 理论计算值与实验值基本一致,不是很接近的原因是计算时没有采用溶剂化作用,但数值在可接受范围。
化合物的DMF溶液(1×10-5 mol/L)的UV光谱图(图5)中,在272和330 nm处出现两个吸收带,前者属于共轭体系的π→π*电子跃迁亦即K带,后者属于n→π*电子跃迁亦即R带。 这些吸收峰分别对应于前线分子轨道的HOMO到LUMO+1的电子跃迁和HOMO到LUMO的电子跃迁。
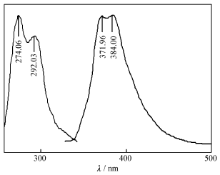
在室温下,化合物的DMF溶液在300~400 nm范围可产生紫色荧光(图6),属于分子的π*→π跃迁。在274 nm波长的光激发下,发射峰的最大发射波长为371和384 nm,分别对应于LUMO+2到HOMO和LUMO到HOMO的电子跃迁。
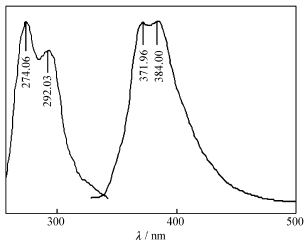 | 图6 H2L的荧光光谱Fig.6 FL spectra of H2L λex=274 nm, λem=371 and 384 nm |
{kind=link}
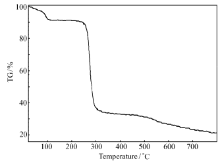
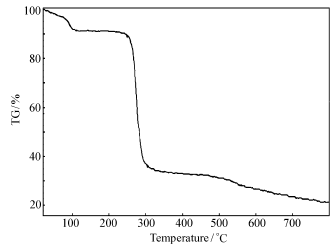 | 图7 标题化合物的热重分析图Fig.7 TGA of title compound |
{kind=link}
分子的总能量为-25339.941 eV,最高占据轨道能量 EHOMO=-5.660 eV,标准焓和标准自由能也均为负值,说明化合物在基态稳定。 热重分析(图7)表明,该化合物在254 ℃以下是稳定的。 室温至100 ℃失去1.5个结晶水,失重8.88%(理论失重9.06%),254 ℃开始分解并快速失重,254~302 ℃失重52.03%,至800 ℃残重21.01%。
测定了2,4-二羟基苯乙酮缩异烟酰腙Schiff碱的晶体结构,晶体属单斜晶系,C2/c空间群,化合物借助分子间氢键形成三维超分子体系。 该化合物有很好的热稳定性和较高的反应活性;在274 nm波长的光激发下,可发射紫色荧光,可用于开发新的发光材料。 化合物的酰腙基团和酚羟基具有很强的给电子能力,是金属离子进攻的位点和发生氧化过程的活性中心。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|