

Suyun GAO, Kulisong HAYIERBIEK, Han ZENG. Performance of Biofuel Cell Based on 4-Mercaptobenzoic Acid Functionalized Nanoparticles Tethered with Glucose Oxidase and Laccase[J]. Chinese Journal of Applied Chemistry, 32(6): 708-719


以4-巯基苯甲酸修饰纳米金粒子作为固酶载体和导电基体构建了新型纳米结构固酶葡萄糖/O2燃料电池,其制备简单,长期使用性能稳定。利用纳米金粒子通过表面修饰基团和酶分子活性中心附近疏水结合位之间的相互作用固定葡萄糖氧化酶(GO x)和漆酶(Lac)分子,分别制备了固酶阳极—4-巯基苯甲酸功能化纳米金粒子固定葡萄糖氧化酶修饰金盘电极GO x/4-MBA@GNP/Au和固酶阴极—4-巯基苯甲酸功能化纳米金粒子固定漆酶修饰金盘电极Lac/4-MBA@GNP/Au。电化学实验结果表明,两种电极在不引入任何外加电子中介的条件下,均可以实现酶活性中心-纳米金粒子之间的直接电子迁移,而且具有较快的催化反应能力(固酶阳极和阴极的转化速率分别为1.3和0.5 s-1;催化葡萄糖氧化和氧气还原的起始电位分别为-0.23和0.76 V)。评估了固酶阳极和阴极组装成的纳米结构固酶葡萄糖/O2燃料电池的能量输出性能。该燃料电池在没有Nafion薄膜和阳极无N2气保护下,开路电压和最大输出能量密度分别可达0.56 V和760.0 μW/cm2,使用一周后输出能量密度仍然可以达到最初值的~88%。进一步测试结果显示,该燃料电池呈现出与游离漆酶类似的pH依赖关系和热稳定性,这些实验结果均暗示:影响整个酶燃料电池性能的关键在于漆酶基阴极催化氧还原的过程。此外,这种燃料电池的性能虽然受到共存干扰物抗坏血酸的影响,但在人类血清中测试结果显示其仍然具有较高的输出能量密度(132.0 μW/cm2,开路电压0.40 V)。本文研究结果给出了设计高性能葡萄糖/O2燃料电池的新思路,同时也为研究固酶燃料电池的构效关系提供了实验依据和有价值的启示。
A novel prototype of nano-structure glucose/O2 biofuel cell was constructed with immobilized enzyme on 4-mercapto benzoic acid(4-MBA) modified gold nanoparticles as electrical medium and enzyme carrier. It features with simple fabrication and favorable long-term usability. Glucose oxidase(GO x) and Laccase(Lac) molecules are tethered steadily to the surface of gold nanoparticles via interaction between modified group of gold nanoparticles and hydrophobic binding-sites in the vicinity of cofactor within enzyme. The as-prepared bioanode and biocathode are fabricated and denoted as GO x/4-MBA@GNP/Au and Lac/4-MBA@GNP/Au, respectively. Electrochemical results indicate that the direct electron transfer occurs between enzyme active sites and gold nanoparticles for both biocathode and bioanode in the absence of any mediator achieves with fast catalytic activity(turn-over frequency of bioanode and biocathode:1.3 and 0.5 s-1, onset potential according to the glucose catalytic oxidation and catalytic reduction of oxygen:-0.23 and 0.76 V). The power out-put performance of nano-structure with entrapped enzyme glucose/O2 biofuel cell was further evaluated after the constitution of cell via connection bioanode and biocathode on the basis of previous results. The results of test show that the open-circuit voltage and the maximum out-put energy density of this biocell amount to 0.56 V and 760.0 μW/cm2 in the absence of Nafion ion-exchange membrane and anode protection gas of N2, respectively. The out-put density of biofuel cell after storage in refrigerator for one week can still retain ~88%of the initial value. Furthermore, this fuel cell shares the similar characteristics of pH dependence and thermal stability to those of free laccase. The key factor contributed to the performance of biofuel cell should be related to the catalytic oxygen reduction process at biocahode. Despite of the apparent influence of concomitant interferent-ascorbic acid on cell performance, this cell still shows superior out-put energy density(132.0 μW/cm2, open circuit voltage:0.40 V) recorded in the circumstance of the human serum. This study may afford a new route to design in high performance glucose/O2 fuel cell and provide experimental basis and valuable enlightenment for the study of relationship between the structure of enzyme based cell and its performance.


酶燃料电池是一种新型且很有竞争力的电化学能量转化装置[1,2]。在酶基阳极中,最常见的是固定葡萄糖氧化酶(GO x)基阳极,以葡萄糖作为底物。文献[3-5]报道的具有最佳催化性能的酶基电极均基于GO x活性中心在电极表面重构,以实现酶活性中心与电极的直接电子导通,但这种方法过程繁琐而且需要使用有毒试剂,成本昂贵,不适于在人体内使用;而酶基阴极因为酶催化氧还原的活化能较高,常常被认为是影响整个酶基燃料电池性能的关键[6,7]。在作为阴极催化剂的酶当中,漆酶(Lac)由于具有相对最高的式电位及很高的催化活力而最受关注[6,7,8,9,10,11,12]。但现有固酶阴极都存在一些问题:许多Lac基电极不能实现酶-电极之间的有效直接电子迁移,需要引入外加电子中介体,导致输出电压明显下降,且影响燃料电池的使用寿命和植入生物体内的潜在应用性[6,7,8]。一些无需中介体而能够实现直接电子迁移的Lac基阴极,由于电子迁移的速率不够快,催化氧还原性能欠佳[9,10];另外一些具有较高催化性能的固酶阴极却因为成本高,对环境不友好且制备过程繁琐,长期使用性能不佳而在应用中受到较大限制[11,12]。当将GO x基阳极和Lac基阴极组装为酶燃料电池时,由于 “燃料穿越”往往导致电池能量输出性能远远低于理论预期,从而需要采用离子交换膜,这增大了电池内阻,导致输出能量的损失[13]。现有的具有较高性能的无隔膜碳纤维表面修饰单壁碳纳米管固酶基燃料电池等[14,15],由于碳纤维/纳米管与酶分子之间的复杂作用,导致电极表面固定的酶分子渐渐失活,因此这种酶燃料电池长期使用性能并不好。
纳米金粒子凭借量子效应,小尺寸效应以及相对于大尺寸金电极具有更好的生物分子相容性,成为近来开发酶燃料电池电极材料的最佳选择[16,17]。只要在纳米金粒子表面修饰适当的可与酶分子活性中心附近化学结合位发生相互作用的化学基团,就不仅能增加载体对酶的担载量,还可能增强酶活性中心-纳米金粒子之间直接电子迁移的能力[17]。文献[17,18,19]研究结果表明,GO x和Lac分子活性中心附近均存在疏水的芳香基团化学结合位,因此可以设想,采用具有特定结构的芳香性基团修饰纳米金粒子表面,就可能在稳定固定GO x和Lac的同时实现两种酶分子和纳米金粒子之间的直接电子迁移。这不仅使电极制备过程得到简化而且可以有效消除燃料穿越带来的能量损失。文献[20]报道的巯基苯甲酸功能化纳米金粒子制备简单且具有良好的生物相容性和导电性,同时其表面修饰的巯基苯甲酸基团易于接近GO x和Lac活性中心附近的疏水结合位,多余的羧基也有可能与酶分子表面的氨基化学键合从而可能达到上述目的。迄今为止,尚未见到巯基苯甲酸功能化纳米金粒子同时固定GO x/Lac基葡萄糖-O2燃料电池性能研究的相关报道。现有的固定酶基纳米金粒子电极,或者因为酶-电极之间的复杂作用,使得电极表面固定酶失活,导致酶电极长期使用性能较差,或因为酶在电极表面的不适宜定位导致酶-电极间电子迁移速率缓慢,氧还原催化性能较差。本文采用对两种酶分子都具有较高亲和力和较大担载量的芳香基团功能化修饰纳米金粒子制备GO x基和Lac基电极,在此基础上制备了一种结构简单、制备方便、无需任何中介体就可实现酶-纳米金粒子之间的快速电子迁移,输出能量密度高,热稳定性强,pH耐受范围宽且长期使用性能稳定的酶燃料电池,虽然该电池对抗坏血酸(AA)的耐受性不很理想,但仍存在较大的性能改善余地。
云芝Lac(Lac from Trametes Versicolor,相对分子质量68000),抗坏血酸氧化酶(AAO x,源自Cucurbita sp.),抗坏血酸(AA),牛血清白蛋白(BSA)及GO x(相对分子质量约50000)购自美国Sigma化学试剂有限公司,使用前无需进一步纯化;四水合四氯金酸,硼氢化钠, N-乙基 -N'-(3-二甲基氨丙基)碳二亚胺(EDC), N-羟基琥珀酰亚胺(NHS),2,2'-连氮-双-(-3-乙基苯并噻唑啉-6-磺酸)-二胺盐(纯度:98.5%,ABTS),二茂铁甲酸(FMCA);聚乙烯亚胺(PEI,相对分子质量7.5×105)以及4-巯基苯甲酸(4-MBA)均购自美国Sigma-Aldrich化学试剂有限公司。葡萄糖和戊二醛购自上海阿拉丁试剂有限公司。冰乙酸、甲醇、乙醚以及配制缓冲液所需的试剂则购自国药集团化学试剂有限公司。所有药品如无特殊说明均为分析纯。实验过程中使用的缓冲溶液为0.2 mol/L的磷酸二氢钾缓冲液(PBS),通过改变磷酸二氢钾和柠檬酸三钠的比例调控溶液的pH值,所有溶液均用Milli-Q超纯水配制。
Bruker Equindx-55型红外光谱仪(德国Bruker公司),KBr压片; U-2810型紫外可见分光光度计(日本岛津公司,比色皿厚度:0.5 cm)和Analyst 800型原子吸收光谱仪(美国Perkin Elmer公司),主机:双光束火焰/石墨炉原子吸收分光光度计,光谱范围190~870 nm用于表征电极表面固定酶分子的稳定性;2K15型高速离心机(德国Sigma公司)用于离心沉降分散于混合溶液中的纳米金粒子;ALV/DLS/SLS-5022F型激光光散射光谱仪(德国ALV公司)改进过,用于测定功能化纳米金粒子的平均粒径,配备ALV-5000型数字时间相关器和He-Ne激光光源,波长632 nm,输出功率22 mW;实验前样品溶液经过除尘(dust free)处理,溶液浓度为0.58 mmol/L。测量温度:(24±0.5) ℃, θ=90°;CHI-1140A型电化学分析仪(上海辰华仪器有限公司),作为参比电极的Ag/AgCl(饱和KCl)电极和作为工作电极的金盘电极(GD,直径1 mm)均购自天津艾达恒晟工贸有限公司,高纯N2气和高纯O2气购自南京特气。工作电极使用前先以3500#砂纸,1.0和0.5 μm氧化铝粉浆抛光,再用丙酮和三次重蒸水超声清洗各2次,每次2 min。
不同平均粒径的4-MBA@GNP按照文献[20]报道的方法制备,步骤简述如下:将四氯金酸和4-MBA以摩尔比1∶3共同溶解于35 mL甲醇/乙酸混合液(体积比6∶1)中,快速搅拌条件下加入含0.3 g NaBH4的15 mL甲醇溶液,加热回流30 min,产生的黑色分散相在冷却到室温后继续搅拌30 min。随后将分散相转移到离心管中,于8000 r/min下离心沉降10 min,得到的黑色固体以乙醚冲洗3次,随后用干燥N2气流吹干待用。
1.3.1 固酶阳极的构筑 构筑固定GO x的功能化纳米金粒子修饰金盘电极之前,将预处理的金盘电极浸泡在含分枝型聚合物PEI(质量分数0.5%)的溶液中6 h便可以获得表面为氨基修饰的金盘电极(记为NH2-PEI@GNP)。然后将氨基功能化的金盘电极浸泡在含EDC(浓度0.1 mol/L),NHS(浓度0.02 mol/L)和4-MBA@GNP(粒径:18 nm,浓度为0.5 g/L)的甲醇-乙酸(体积比为10∶1)混合溶液中1 h以获得纳米金粒子修饰电极(记为4-MBA@GNP/Au)。最后将纳米金粒子修饰电极浸泡入含GO x浓度为3.0 g/L的0.2 mol/L PBS缓冲溶液中(pH=6.0),于4 ℃冰箱中静置过夜,可制备得到固定GO x的纳米结构阳极(记为GO x/4-MBA@GNP/Au),电极不使用时浸泡在前述PBS缓冲液中于4 ℃冰箱中保存。电极使用时先用PBS缓冲液缓慢且充分地润洗2次以除去表面固定不紧密的GO x分子,然后在室温常压下使用。
1.3. 2固酶阴极的构筑 按照1.3.1节相同的方法构筑固定Lac基阴极,只是将加入PBS缓冲溶液中的酶换为Lac,制备的电极记为Lac/4-MBA@GNP/Au。为了保证实验结果的重现性,每次电化学测试时需要制备5个酶电极(阳极和阴极皆如此)。为了测试固酶阴极的稳定性,固酶电极在含ABTS为0.5 mmol/L的PBS缓冲液中静置14 d,测定溶液在420 nm处吸光度与时间的关系曲线,也可使用石墨炉原子吸收法测定溶液中铜浓度的变化(1个漆酶分子含有4个铜离子)[21]。
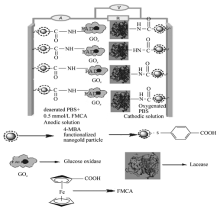
将构筑的固定GO x的阳极GO x/4-MBA@GNP/Au和固定Lac的阴极Lac/4-MBA@GNP/Au,放入一个玻璃制两极室电池中,以全氟化离子交换膜(Nafion 115,厚度0.125 mm)作为隔膜将阳极室和阴极室(体积各为10 mL)分隔开。两个极室上方各有一个进气口,以导气管与气源连接,阳极电解液含有不同浓度的除氧葡萄糖PBS缓冲溶液(pH=6.0)。为了提高燃料电池输出功率,需要改善阳极GO x与电极之间的电子迁移性能,因此在阳极电极液中加入FMCA作为电子中介体,其浓度恒定为0.5 mmol/L;而阴极室则充入为氧气饱和的PBS缓冲溶液(pH=6.0),不需加入任何电子中介体。电阻值范围在0~100 kΩ的外加负载与组装的两个电极相连接,籍调节负载的电阻值调控电池的输出电压和输出电流密度。电池的结构及组装示意图参见Scheme 1。
 | Scheme 1 Schematic representation of the GO x-Laccase based biofuel cell |
{kind=link}
固酶阳极GO x/4-MBA@GNP/Au和固酶阴极Lac/4-MBA@GNP/Au分别作为工作电极,与参比电极(饱和KCl作为电解质的Ag|AgCl电极)和对电极(铂丝)连接成一个三电极系统,置于一个容积为25 mL的玻璃电解池中,测试固酶阳极直接电化学性质前,电解池内注入不同葡萄糖浓度的PBS缓冲液并以高纯N2气鼓泡除氧至少30 min,测试时电解液上方继续通入N2气以维持无氧气氛;而测试固酶阴极直接电化学性质前,电极中的PBS缓冲液先以高纯N2气鼓泡除氧至少30 min,在电解液上方维持N2气气氛的条件下测试固酶阴极的直接电化学行为,随后向PBS缓冲液鼓泡通入高纯氧气至少15 min以使溶液为氧气饱和,在同样条件下测定固酶阴极对氧还原的催化性能。
本文中如无特殊说明,所牵涉的电位均为相对于AgCl|Ag(饱和KCl)参比电极的值。酶基阳极和阴极的活性表面积按文献[7]给出的方法以铁氰化钾为探针测定,分别为0.009和0.01 cm2。测定的电流密度系输出电流对活性面积归一化所得。文中给出的电流-电压曲线( i-E曲线)均为电极达到稳定状态时扫描所得(即第5圈扫描时所得 i-E曲线)。为了消除环境中存在的AA对制备的燃料电池电极输出性能的影响,在测定燃料电池性能之前需将酶基阳极和阴极分别浸入含有BSA(质量分数为1.5%),戊二醛(质量分数为1.0%)和AAO x(活力为1.2 U/μL)的混合液中30 min,在室温空气中干燥,再以重蒸水润洗两次,室温下干燥后待用。使用的人类血清来自健康志愿者提供血样,采集血样后立即在8000 r/min下离心20 min获得测试所需血清,采集血样1 h后,将以氧气饱和的人类血清作为电解质溶液(其中FMCA浓度为0.5 mmol/L),进行酶基燃料电池性能测试。所有实验操作皆在室温((25.0±0.40) ℃),常压下进行。
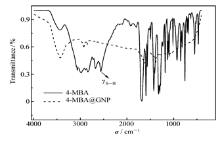
将4-MBA@GNP与4-MBA的FTIR进行比较(图1)后发现:4-MBA@GNP的FTIR中2556 cm-1处对应于4-MBA中S—H伸缩振动吸收峰 γS—H消失了,而其它吸收峰的位置与4-MBA的FTIR相似,这个结果与文献[20]报道相似,表明4-MBA已经成功修饰到纳米金粒子的表面,4-MBA通过Au—S键与纳米金粒子结合。制备的4-MBA@GNP以激光光散射光谱仪测定它们的平均粒径分别为10、18、40和110 nm。固定Lac阴极的稳定性测试结果表明,当固酶电极在含ABTS的PBS中静置14 d后,溶液的吸光度基本不随时间发生变化,同时石墨炉原子吸收法测定结果也表明电极在室温下或溶液温度升到345 K时,溶液中铜浓度均无显著的增加,以上实验结果表明,4-MBA@GNP已经稳定地固定了Lac分子。此外以铁氰化钾作为探针,金盘电极和固定纳米金粒子的金盘电极在同一溶液中得到的CV有明显差别,后者峰电位差更小,峰电流更大,PEI-NH2修饰的金盘电极与金盘电极相比则出现峰电流降低,峰电位差增加的现象(与文献[13,21]报道结果类似,这里不再给出)。上述旁证表明,只有当电极表面修饰有PEI的氨基时,才可以进一步通过偶联功能化纳米金粒子稳定锚接酶分子,而当NH2修饰金盘电极在固定纳米金粒子前后,电极导电性能存在明显差异。对于固酶阳极也可以使用类似的方法研究其表面固定酶分子的稳定性,获得了类似的结果(结果不再赘述)。
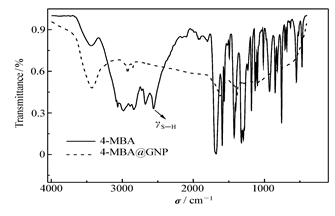 | 图1 4-MBA@GNP和4-MBA的FTIR光谱Fig.1 FTIR spectra of 4-MBA@GNP and 4-MBA |
{kind=link}
图2为GO x/4-MBA@GNP/Au在不含底物葡萄糖的无氧PBS(pH=6.0)中的循环伏安曲线(CV)。从图2可以看出,当PBS缓冲液中既不含底物葡萄糖也没有氧气和电子中介体时,获得的CV曲线上只能看到一对非常图明显的葡萄糖氧化酶活性位核黄素腺嘌呤二核苷酸(FAD)准可逆氧化还原峰(氧化峰电流 ip,a/还原峰电流 ip,c=0.8,氧化峰峰电位-413 mV,还原峰峰电位-494 mV),中值电位-444 mV,很接近文献[21]报道的葡萄糖氧化酶活性中心FAD的式电位-220 mV vs. NHE)。由于纳米金粒子与葡萄糖氧化酶活性中心FAD分子的复杂作用(例如金与含氮杂环之间的络合作用以及氢键,静电吸引等作用)可能导致FAD自身氧化受到抑制,因此造成阴阳极峰电流相差较大,可逆性较文献[7]报道的羟基磷灰石-多壁碳纳米管复合物固定葡萄糖氧化酶基电极相对差一些,但峰电位差(81 mV)与之相近(80 mV);作为对比,没有固定GO x的纳米金粒子修饰金盘电极在同样的条件下测试则没有氧化还原峰,这进一步表明固定在电极表面纳米金粒子上的GO x实现了和导电基体之间的直接电子迁移。此外按文献[4]介绍的方法求出电极表面导电的GO x表面浓度为1.3×10-8mol/cm2,此值与文献[7]报道的碳纳米管-羟基磷灰石复合物固定的导电GO x表面浓度(1.5×10-8 mol/cm2)非常接近。
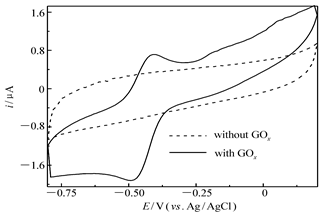 | 图2 GO x/4-MBA@GNP/Au在0.2 mol/L无氧PBS(pH=6.0)中的循环伏安曲线scan rate:10 mV/sFig.2 Cyclic voltammogram of GO x/4-MBA@GNP/Au in 0.2 mol/L deaerated PBS(pH=6.0) |
{kind=link}
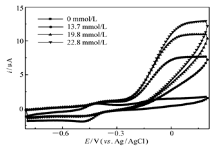
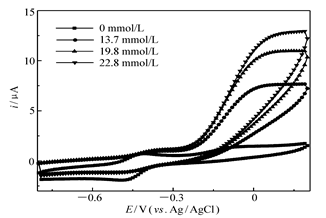 | 图3 GO x/4-MBA@GNP/Au在含不同浓度葡萄糖的0.2 mol/L无氧PBS(pH=6.0)中的循环伏安曲线Fig.3 Cyclic voltammograms of GO x/4-MBA@GNP/Au in 0.2 mol/L deaerated PBS(pH=6.0) with variable concentrations of glucosescan rate:10 mV/s |
{kind=link}
图3为固酶电极GO x/4-MBA@GNP/Au在含不同浓度葡萄糖的0.2 mol/L无氧PBS中扫描所得的CV。从图3可以看出,当缓冲液PBS中加入葡萄糖后,在-0.23 V处开始氧化电流有明显的增加,在0 V左右氧化电流趋于水平的同时伴随着阴极峰电流的降低,而且随着加入葡萄糖浓度的升高,极限氧化电流的数值也相应地增加。这些现象都说明固定在阳极上的GO x不但在无底物时可以从导电基体上以准可逆方式得失电子,同时也可以有效地催化底物葡萄糖的氧化(相对于文献[7]报道的使用了中介体FMCA体系(其起始氧化电位约为0.24 V),本文制备的固酶阳极不使用中介体。而且起始氧化电位负移了约470 mV)。此外,根据加入葡萄糖浓度足够高(葡萄糖浓度高于60 mmol/L)时测定得到的极限催化电流(31 μA)和前述计算得到的导电酶分子的表面浓度,可以估算出固酶催化底物分子反应速率为1.3 s-1,这一数值与葡萄糖-氧气分子化学反应速率[8](600 s-1)相比非常小,因此溶液中含有的氧气可能会影响阳极的功率输出,这也暗示组装成燃料电池时,阳极室和阴极室可能需要使用隔膜分隔开以避免“燃料穿越”带来的能量损失。由于Lac与导电基体之间的直接电子迁移性能对于燃料电池阴极的能量输出大小有着至关重要的作用,因此提高阴极固定的Lac催化氧气分子还原成水的速率就成为改善燃料电池阴极性能的必然选择。
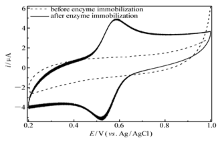
图4给出了Lac/4-MBA@GNP/Au在不含底物氧气和任何中介体的PBS(pH=6.0)中的循环伏安曲线(CV)。从图4可以看出,作为对比的没有固定Lac的4-MBA@GNP/Au在扫描电位范围内没有出现任何氧化还原峰,
 | 图4 Lac/4-MBA@GNP/Au在0.2 mol/L无氧PBS(pH=6.0)中的循环伏安曲线Fig.4 Cyclic voltammogram of Lac/4-MBA@GNP/Au in 0.2 mol/L deaerated PBS(pH=6.0)scan rate:100 mV/s |
{kind=link}
这表明该电极并不含在此电位区间内可以发生氧化或还原反应的基团;固酶电极在相同的电位扫描区间内却出现了1对氧化还原信号,其中值电位为552 mV(氧化峰585 mV,还原峰518 mV,峰电位差为67 mV阴阳极峰电流之比 ip,a/ ip,c=0.86),根据上述分析结果和文献[22]报道的Lac各个活性中心的中值电位数据,这对氧化还原峰可归因于电极表面固定的Lac的活性中心T1(中值电位 E1/2=780 mV vs.NHE)与导电基体之间发生单电子的准可逆氧化还原反应。
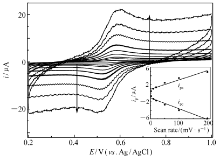
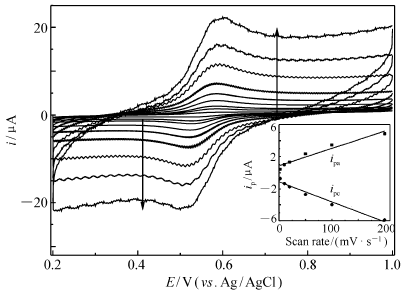 | 图5 Lac/4-MBA@GNP/Au在0.2 mol/L无氧PBS(pH=6.0)中以不同扫速扫描所得循环伏安曲线Fig.5 Cyclic voltammograms of Lac/4-MBA@GNP/Au in 0.2 mol/L deaerated PBS(pH=6.0) with different scan ratesfrom inner to outer CVs corresponding to scan rate:2, 10, 20, 50, 100, 200, 500, 1000, 2000 mV/s, inset:plots of anodic and cathodic peak currents versus scan rates |
{kind=link}
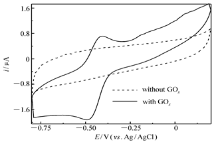
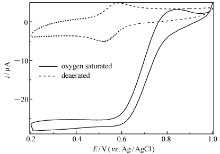
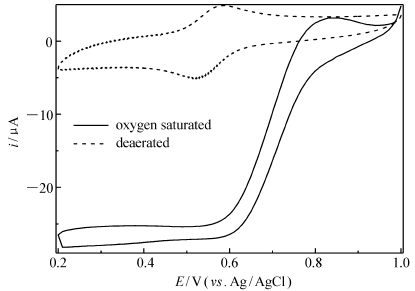 | 图6 Lac/4-MBA@GNP/Au在无氧(虚线)和氧气饱和(实线)0.2 mol/L PBS(pH=6.0)中的循环伏安曲线Fig.6 Cyclic voltammograms of Lac/4-MBA@GNP/Au in deaerated(dash line) and oxygen saturated(solid line) 0.2 mol/L PBS(pH=6.0) at scan rate 100 mV/s |
{kind=link}
而根据前述估算方法可以类似的估算出阴极表面导电的Lac分子表面浓度为1.24×10-8 mol/cm2,这个数值大大高于Lac分子在载体表面紧密单分子层排列的表面浓度4.3×10-12 mol/cm2,也远高于文献[23]报道的多孔纳米金固酶分子表面浓度(2.1×10-11 mol/cm2),接近于文献[24]报道的NAFION薄膜吸附漆酶修饰热解石墨电极上导电酶分子表面浓度(6.6×10-8 mol/cm2)。图5则进一步给出了该固酶电极在相同体系中不同扫速的CVs。从图5可以看出,无论是对于阳极峰电流还是阴极峰电流,在测试的低扫速范围(2~200 mV/s)内,峰电流和扫速一次方保持良好的线性关系(拟合所得的线性相关系数分别是0.995和0.992),而且峰电位基本保持不变,阴阳极峰电流之比接近于1,这进一步证实发生在电极表面上的电化学反应是一个表面控制型的准可逆单电子氧化还原反应。图6为该固酶电极分别在氧气饱和和无氧的不含任何中介体的PBS(pH=6.0)中以相同电位扫描速率获得的CVs。从图6可以明显看出,两种不同条件(无氧和氧气饱和)下的CVs存在明显不同:当溶液为氧气饱和时,固酶电极Lac/4-MBA@GNP/Au的CV中阴极电流在0.76 V开始急剧增大并在0.6 V附近达到极限(电位继续负移,电流变化相对变化较小),同时阳极氧化峰(相对于无氧时测得的CV)消失,这和文献[14]报道的现象相一致,表明电极固定的Lac不但实现了酶活性中心和导电基体之间的直接电子迁移并且有效催化了氧气分子的电还原,氧还原超电势仅为75 mV,这个数值远低于文献[23]报道的多孔纳米金固定Lac电极催化氧的超电势(400 mV),甚至比最近文献[11]报道的2-氨基蒽醌修饰单壁碳纳米管表面吸附的诱陷Lac的NAFION层基电极的氧还原超电势(122 mV)还低将近50 mV。而根据0.4 V时的极限催化电流 icatlim=23.8 μA以及前面计算得出的导电酶分子表面浓度 Γ、电极活性表面积等数据,借助文献[17]给出的方法估算出实现直接电子迁移的Lac分子单位时间内转化氧分子的频率为0.5 s-1,这个数值不到前面计算得到的GO x催化葡萄糖氧化速率(1.3 s-1)的一半,说明阴极动力学可能成为制约燃料电池输出功率的主要因素,虽然这个速率较文献[11,12,23]报道的导电酶分子催化氧还原速率(分别是0.14、0.09和0.06 s-1)要高得多。
一般而言,为了避免燃料“穿越”带来的电池输出能量损失,需要以Nafion隔膜将燃料电池两极室分隔开,但加入离子交换膜会增大电池内阻造成能量损失,如果酶-电极间电子迁移速率快到足以克服燃料“穿越”的负面影响的时候,这个薄膜就不再成为必需。
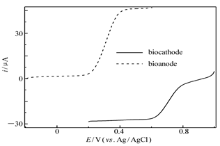
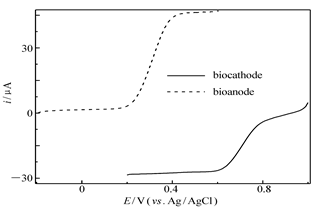 | 图7 酶基阳极GO x/4-MBA@GNP/Au(虚线)在含1.0 mmol/L葡萄糖和0.5 mmol/L FMCA的无氧0.2 mol/L PBS(pH=6.0)中和酶基阴极Lac/4-MBA@GNP/Au(实线)在O2饱和0.2 mol/L PBS(pH=6.0)中的极化曲线Fig.7 Polarization curves obtained in 0.2 mol/L PBS for the bioanode GO x/4-MBA@GNP/Au(dash line) in deaerated solution contains 1 mmol/L glucose+0.5 mmol/L FMCA and for the biocathode Lac/4-MBA@GNP/Au(solid line) in oxygen-saturated solution recorded at scan rate:100 mV/s |
{kind=link}
此外如2.1节所述,固酶阳极在没有电子中介体存在的情况下,虽然可以实现酶活性中心与导电基体-功能化纳米金粒子之间的直接电子迁移,但迁移速率不够快,产生的催化电流密度也不够高,因此为了提高阳极的输出功率往往需要使用电子中介体,而目前最常使用同时也是催化葡萄糖氧化效率最高的阳极中介体是FMCA[7,21]。图7为固酶阳极GO x/4-MBA@GNP/Au在含葡萄糖和FMCA的无氧PBS(pH=6.0)以及固酶阴极Lac/4-MBA@GNP/Au在O2饱和的PBS(pH=6.0)中以相同电位扫描速率获得的极化曲线。从图7可以看出,固酶阳极催化葡萄糖氧化始于约200 mV处,而固酶阴极催化氧还原的起始电位在760 mV左右,当反应由底物(葡萄糖和氧气)扩散控制时,催化电流趋于稳定,即不再随电位变化而有明显的改变。值得注意的是,当阴阳极底物浓度接近时(溶液中溶解氧气的浓度大约为1.2 mmol/L),阳极极限催化氧化电流要高出阴极极限催化还原电流约1/3,这一结果与文献[7]报道的碳纳米管-羟基磷灰石固酶燃料电池测定结果类似,但后者阴阳极极限催化电流相差更大(相差将近1倍),这表明反应是由底物氧气和葡萄糖的传质扩散控制,而且还暗示电池的能量输出受限于阴极氧分子催化还原过程,这可能源于固酶载体对氧分子的亲和力,氧分子在固酶载体上化学吸附的速率以及氧分子在固酶载体表面结合位和直接电子迁移发生位点之间的距离[7,24]。
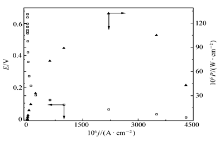
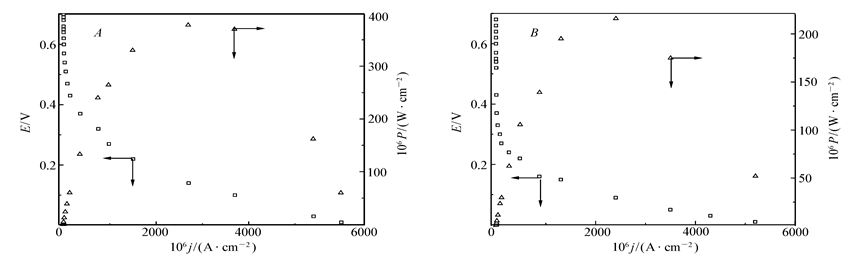
图8分别给出了4-MBA@GNP固酶基葡萄糖/O2生物燃料电池在两种不同情形下于特定测试体系中得到的极化和性能曲线:有Nafion薄膜分隔开阳极室和阴极室且阳极室处于N2气气氛(图8 A)以及没有隔膜存在且阳极不再充入N2气保护(图8 B)。从图8 A图可以看出,固酶燃料电池的开路电压(OCV)约为0.66 V,在0.31 V处呈现的最大输出能量密度大约是840.0 μW/cm2。由于酶-电极间迁移速率不够快以及存在隔膜电阻等因素的综合影响,尽管OCV(0.66 V)相对于理论上此电池最大输出电压0.98 V仍然存在320 mV左右的电压差,但与文献[7]报道的羟基磷灰石-多壁碳管固酶基燃料电池的OCV(0.58 V)和最大输出电流密度(0.28 V时的最大能量密度为15.8 μW/cm2)以及碳纳米管基葡萄糖/AA-O2酶燃料电池[15]的性能(OCV 0.62~0.84 V,但其最佳输出能量密度仅为9.5~10 μW/cm2)相比仍然具有相当的优势,由于本文制备的电极以功能化纳米金粒子作为导电基体,相对于多壁碳管对底物和中介体的传质性能更好[10,16]。纳米金粒子表面修饰的4-MBA含有疏水的苯环可以凭借疏水-疏水分子间作用力,选择性与Lac分子和GO x分子活性位附近的疏水结合位发生作用而紧密吸附,利于酶活性中心和纳米金粒子之间的电子迁移,因此产生更高的催化电流密度和较小的反应超电势。从图8 B图则可以进一步看出,当薄膜不存在且阳极不再充入保护气时,固酶燃料电池的OCV下降到了560 mV,但在0.39 V处的最大输出能量密度仍然可以保持为760.0 μW/cm2。这个实验结果表明,当隔膜不存在且无保护性气体时,电池内部虽然也会发生燃料“穿越”造成能量的下降,但只损失了不到15%的能量,且此情况下获得的最大输出能量密度仍然远高于文献[7]报道的相似固酶电极。综上所述,本文制备的纳米金粒子固酶基燃料电池具有较快的酶-导电基体间电子迁移速率和较好的催化特性,可以在不使用离子交换膜和不充入阳极保护气条件下工作。
 | 图8 4-MBA@GNP固酶基葡萄糖/O2生物燃料电池的极化和性能曲线Fig.8 Polarization curves(filled square) and performance curves(empty triangle) of as-assembled 4-MBA@GNP with immobilized enzyme based glucose/O2 biofuel cell A.in the presence of Nafion membrane and anolyte bubbled with N2, catholyte saturated with O2; B.Nafion separator-free and catholyte:PBS saturated with O2, anolyte: oxygen saturated PBS contains 1 mmol/L glucose+0.5 mmol/L FMCA |
{kind=link}
因为纳米金粒子的粒径对于固酶电极的催化性能有不可忽略的影响,所以在测试酶基燃料电池性能之前,需要研究纳米金粒子的粒径和电池性能之间的关系,以确定最合适的纳米金粒子的粒径。在图8 B对应的测试条件下,研究了纳米金粒子粒径分别为10、18、40和110 nm时的酶基燃料电池性能,结果表明,纳米金粒子的粒径变化对燃料电池OCV和最大功率对应电压影响不大,OCV在0.55~0.57 V之间变动,而最大功率对应电压基本维持在~0.39 V左右,粒径为18 nm时有最大输出功率(粒径分别为10、18、40和110 nm时的酶基燃料电池最大输出功率分别为420、760、550和215 μW/cm2)。这一结果与文献[23]报道结论类似,表明当纳米金粒子过大时由于比表面积下降,降低了固载酶分子的容量,而且其表面固定的酶也更容易凭借分子间作用力结合形成团簇状物质使其对底物结合能力下降,降低了电池输出功率。
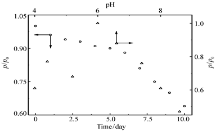
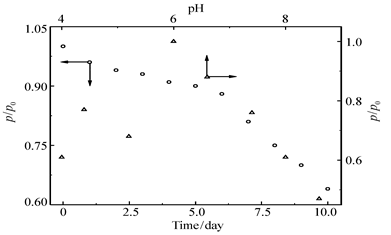 | 图9 无膜固酶葡萄糖/O2生物燃料电池的长期使用性和pH稳定性Fig.9 Long-term reliability and pH stability of glucose/O2 biofuel cell with immobilized enzyme in the absence of Nafion membrane P0:initial maximum power density, other conditions are the same as those inFig.8 |
{kind=link}
图9为4-MBA@GNP固酶基葡萄糖/O2生物燃料电池的性能与工作时间/溶液pH值之间的关系曲线。从图9可以看出,在固酶燃料电池工作的最初6 d内,电池性能下降速率较慢,使用时间超过一周后下降速率急剧增加,使用一周后输出能量密度仍然可以达到最初值的约88%,此酶燃料电池的长期使用性能远优于文献[11]报道的单壁碳纳米管固定Lac基电极燃料电池 (使用6 h后输出功率就降低到初始值的约20%)和碳纳米管基葡萄糖-O2酶燃料电池[15](使用3 d后酶燃料电池的输出能量约是初始值的63.6%),这表明固酶电极在保持较高酶容量的同时,也有效地保持了酶的固有催化活力;此外从图9还可以看出,固酶燃料电池的输出能量随溶液pH值变化的关系与游离Lac催化活力-pH关系[21]相类似,即溶液pH值在4.0~8.6之间时输出能量密度出现两个极值(在pH值为4.4和6.0时,输出能量密度可达585和760 μW/cm2),当溶液pH值超过6.0后输出能量密度迅速下降,当溶液pH值接近弱碱性(pH=8.6)时,输出能量已经降到不到最大输出能量密度的一半。根据上述测试结果可以合理推测,此电池性能受控于固定漆酶基阴极的催化过程,本文制备的酶基电极对pH耐受性较文献[25]报道的Lac基电极更高(该电极在pH=6.0时催化活力已经下降到不到初始值的1/4)。
图10给出了阴阳极均固定一层AAO x的无膜固酶葡萄糖/O2生物燃料电池分别在不含AA(图10 A)和含80 μmol/L AA(图10 B)的电解质溶液中的极化曲线和能量密度-电流密度关系曲线。从图10可以看出,在静态条件下该燃料电池在不含AA的电解质溶液中工作时,测得的OCV约为0.56 V(与无离子交换膜且表面没有固定AAO x时测得的OCV相比几乎没有改变),而在0.14 V时的最大输出功率约为380 μW/cm2,此数值与图8 B获得的测试结果相比仅为后者的一半,表明表面固定的AAO x可能阻碍了葡萄糖或氧分子与对应酶分子的接触导致输出电流密度下降;而当此燃料电池在含AA的溶液中工作时,测得的OCV则下降到了0.48 V,最大输出电流密度进一步下降到了216 μW/cm2,这与文献[15]报道的类似体系有明显的差别,后者使用的固酶的单壁碳纳米管修饰碳纤维电极,在表面固定一层AAO x之后虽然可以有效排除AA对电池性能的干扰,其OCV和最大输出能量密度均几乎不变,但其获得的最大电流密度(与本文测定时AA的浓度相同)也只有本文报道的1/4,这个测试结果表明,虽然本文制备的电极不能充分排除AA的干扰,但是由于电极表面固定的酶对底物有较高的亲和力,酶-导电基体间电子迁移速率也足够快,因此仍然能够产生足够高的输出能量密度。
 | 图10 阴阳极都固定了AAO x的无膜固酶葡萄糖/O2生物燃料电池对AA的耐受性Fig.10 Endurance of glucose/O2 biofuel cell with immobilized enzyme in the absence of Nafion membrane on the interferent of AA with the immobilization of AAO x onto both the bioanode and biocathode |
{kind=link}
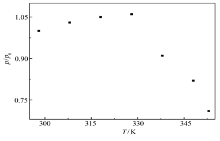
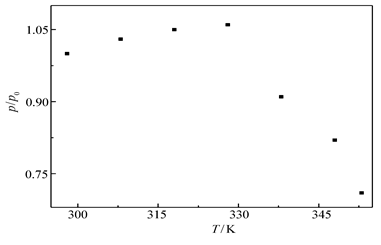
图11为4-MBA@GNP固酶基葡萄糖/O2生物燃料电池的最大输出能量密度和温度的依赖关系曲线。从图11可以看出,在测试温度范围内,随着温度升高,在328 K之前最大输出能量密度缓慢线性升高,而当温度超过328 K之后输出功率急剧下降,这个结果与文献[21]报道固酶电极的催化电流密度-温度关系非常相似,这表明此燃料电池的性能很大程度上受控于Lac基阴极的动力学过程,电极上修饰的纳米金粒子以化学吸附的形式固定酶,当温度升高后酶变性从而造成电池输出性能下降。此外根据图中数据可以估算出电池催化反应的表观活化能约为2.26 kJ/mol,而酶变性的活化能则约为16.4 kJ/mol。
 | 图11 无膜固酶葡萄糖/O2生物燃料电池的热稳定性Fig.11 Thermal stability of glucose/O2 biofuel cell with immobilized enzyme in the absence of Nafion membrane P0:initial power density obtained under the room temperature |
{kind=link}
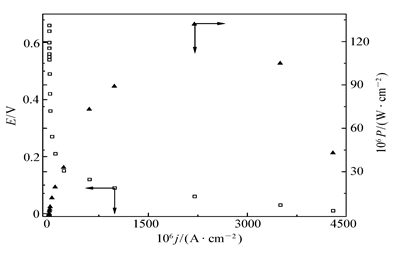 | 图12 无膜固酶葡萄糖/O2生物燃料电池(阴阳极均固定AAO x时)在人类血清中测试得到的极化曲线和输出能量密度-电流密度关系曲线Fig.12 Polarization curve and the relationship between the power output density and the current density of glucose/O2 biofuel cell with immobilized enzyme and the immobilization of AAO x onto both the bioanode and biocathode in the absence of Nafion membrane in human serumHuman serum saturated with oxygen and concentration of FMCA at 0.5 mmol/L, data recorded at room temperature and normal atmosphere pressure |
{kind=link}
图12给出了阴阳极表面都固定了一薄层AAO x的无膜固酶基燃料电池在人类血清中测试得到的极化曲线和输出能量密度-电流密度关系曲线。从图12可以看出,当该电池于人类血清中工作时,其OCV(0.40 V)和最大输出能量密度(132 μW/cm2)与图10 B所示的相同电池在人为控制干扰物质AA浓度一定的体系中测定结果相比,有明显的下降,但仍然比文献[15]报道的固酶单壁碳纳米管修饰碳纤维电极基葡萄糖/O2在类似体系中获得的最大功率密度~35 μW/cm2高得多。这一结果与图10分析结论相一致,即表面固定的一薄层AAO x并未能有效排除干扰物质的干扰而且可能阻碍了固定的酶分子与底物的接触,或者是妨碍了GO x和Lac分子与纳米金粒子之间的直接电子迁移,这表明需要对纳米金粒子表面修饰基团和固定酶分子之间的作用进行深入研究,以制备既能有效排除干扰物质,又不妨碍电极表面固定酶催化剂与导电基体之间的电子迁移以及和底物分子的有效接触的高效酶基电极。
使用4-MBA@GNP作为导电基体和固酶载体,分别制备了GO x和Lac基燃料电池的阳极和阴极,在此基础上组装了以葡萄糖和氧气为燃料和氧化剂的酶基燃料电池。电化学测试结果表明,固定在纳米金粒子上的酶分子(无论是GO x还是Lac),在没有电子中介体的情况下,可以实现酶活性中心和导电基体之间的直接电子迁移,而且,在无离子交换膜且可能存在燃料“穿越”的情况下,酶燃料电池的输出能量密度仍可以保持最大值的85%。进一步测试表明,该酶燃料电池具有良好的热稳定性、长期使用性和pH耐受性,该电池性能受制于阴极氧分子还原过程,此外,这种电池在人类血清中测试也具有较高的输出能量密度(132 μW/cm2)。这种酶燃料电池性能测试结果表明,导电的固酶载体对酶的亲和力以及酶-固酶载体表面修饰基团之间的相互作用,对载体固酶容量和酶-导电基体间的直接电子迁移能力有重要影响,这些结果为设计新型高效酶燃料电池和研究酶燃料电池的构效关系提供了实验依据和新思路、新策略。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|
| [17] |
|
| [18] |
|
| [19] |
|
| [20] |
|
| [21] |
|
| [22] |
|
| [23] |
|
| [24] |
|
| [25] |
|