

Dechun ZHANG, Wen'ge WANG, Aiming SUN, et al. Synthesis and UV Spectra of Aromatic Oligosulfonate-bridged Macrocycles[J]. Chinese Journal of Applied Chemistry, 32(3): 292-298


以芳香二磺酰氯和芳香二酚为原料,三乙胺作缚酸剂,二氯甲烷作溶剂,采取一步成环法合成了一系列新型的、芳环单元不同的芳香寡聚磺酸酯桥联大环化合物。 合成产物的结构用IR、1H NMR、13C NMR和MALDI-TOF等技术手段进行了确认。 并对合成方法、紫外光谱和变温核磁结构研究进行了有价值的探讨。 化合物3~5的DMF溶液的最大紫外吸收峰在266 nm处,化合物6在267 nm处,摩尔吸光系数分别是4.65×104、5.61×104、5.09×104和9.98×104 L/(mol·cm);检出限分别是2.15×10-7、1.78×10-7、1.96×10-7和1.00×10-7 mol/L。 变温核磁数据证明,苯环上连有支链有利于固定构象,可以通过这种方法合成结构固定的杯芳烃。
A series of aromatic oligosulfonate-bridged macrocyclic compounds was synthesized using aromatic disulfonyl chlorides and aromatic diphenol as the starting materials. The products were characterized by IR,1H NMR,13C NMR and MALDI-TOF-MS. The synthetic methods, UV spectra and variable-temperature1H NMR experiments were also discussed herein. UV spectra maximum absorption peaks of compounds 3~5 and compound 6 in DMF solutions are at 266 nm and 267 nm, respectively. The molar absorption coefficients are 4.65×104, 5.61×104, 5.09×104 and 9.98×104 L/(mol·cm), respectively. The detection limits are 2.15×10-7, 1.78×10-7, 1.96×10-7 and 1.00×10-7 mol/L, respectively. Variable-temperature1H NMR experiments show that branched benzene rings can lock the conformation and facilitate the synthesis of desired macrocylces.

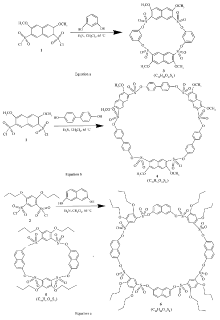
芳香大环化合物可作为一个有效的合成平台,通过芳环上下缘结构修饰和骨架桥联部分杂原子化的建构,引入各种官能团,可形成结构和性能较为独特的三维空间,从而可以借助氢键、静电作用和范德华力等对金属阳离子、阴离子和有机分子产生选择性的识别能力[1,2]。
含氮、硫等杂原子的官能团的引入可以很好的增强对金属阳离子的配位能力[3,4,5,6,7]。 目前,含酰胺基[8,9]、磺酰胺基[10]和巯基[11]等官能团的大环化合物的合成和识别有见报道。 我们将磺酸酯基引入到桥联部分,构建了结构独特、可多点络合和刚性的大环化合物[12,13]。 我们曾报道了具有类似于杯芳烃锥式构象的芳香寡聚磺酰胺大环的一步环化合成[10,14,15]。
本文以芳香二磺酰氯和芳香二酚为原料,三乙胺作缚酸剂,二氯甲烷为溶剂,采取一步成环法合成了磺酸酯桥联杯大环化合物3~6,并得到了IR、1H NMR、13C NMR和MALDI-TOF等对其结构的确认。 将磺酸酯基引入桥联部分,是对杯芳烃基本框架结构进行的改造,具有非常重要意义(合成路线如Scheme 1所示)。
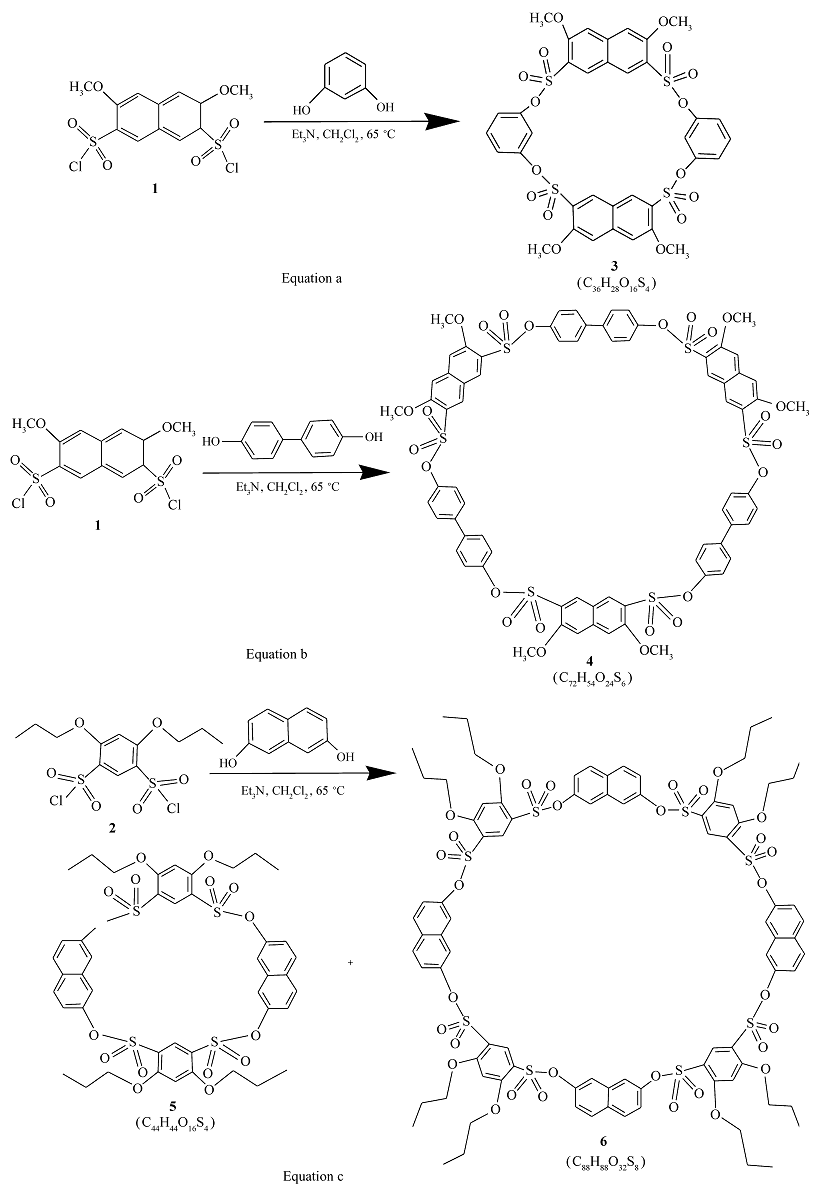 | Scheme 1 Synthesis of the sulfonate-bridged macrocyclic compounds 3~6 |
{kind=link}
Avance Brucker-500 MHz型核磁共振仪(1H NMR,德国Brucker公司);Bruker Daltonics Inc. BIFLEX III型MALDI-TOF质谱仪(德国Brucker公司);Nicolet iS10 FT-IR型红外光谱仪(美国Nicolet公司),KBr压片;UV-2700型紫外光谱仪(日本岛津公司);SLRD-I型数字熔点测定仪(南京桑力电子设备厂)。
间苯二酚、2,7-二羟基萘、4,4'-二羟基联苯二酚、石油醚、丙酮、乙酸乙酯等均为分析纯试剂,溶剂经干燥处理后使用。 化合物3,6-二甲氧基-2,7-萘二磺酰氯(1)[10]和4,6-二正丙氧基-1,3-苯二磺酰氯(2)[14]按文献方法合成。
化合物3,向100 mL的两口瓶中依次加入1.155 g(3 mmol)3,6-二甲氧基-2,7-萘二磺酰氯1的二氯甲烷(10 mL)溶液,0.330 g(3 mmol)间苯二酚的二氯甲烷(10 mL)溶液,0.81 mL(6 mmol)三乙胺,油浴65 ℃回流17 h。 TLC跟踪原料点消失,结束反应。 蒸出溶剂,得粗产品。 上硅胶柱层析,以 V(甲醇)∶ V(乙酸乙酯)∶ V(甲酸)=5∶50∶3的混合溶剂洗脱后,再在DMF中重结晶干燥,得白色晶体0.788 g,产率为62.3%,mp>356 ℃;IR(KBr), σ/cm-1 :1056,1103,1176,1220,1249,1461,1491,1596,1618,1712,2922,3109;MS(MALDI-TOF) m/z:867.3(M++Na)(C36H28O16S4,FW 844);1H NMR(500 MHz,DMSO-d6), δ:4.15(s,12H),6.85(d, J=7.5 Hz,4H),7.12(t, J=7.9 Hz,2H),7.34(s,2H) ,7.67(s,4H),8.28(s,4H);13C NMR(125 MHz,DMSO-d6):57.4,107.9,117.9,119.8,120.2,122.2,131.6,135.9,142.8,149.8,156.2 ;元素分析(按C36H28O16S4·H20计算值)/%:N 0.02(0.00),C 50.21(50.12),H 3.75(3.48)。
用4,4-二羟基联苯二酚等代替间苯二酚,按同方法制得化合物4。
化合物4,白色晶体0.594 g,产率为26.5%,mp 300~301 ℃;IR(KBr), σ/cm-1 :1055,1149,1176,1196,1465,1490,1619,1653,2922;MS(MALDI-TOF) m/z:1516.7(M++Na),1532.7(M++K)(C72H54O24S6,FW 1494);1H NMR(500 MHz,CDCl3), δ:4.11(s,18H),7.11(d, J=7.7 Hz,12H),7.53(d, J=7.8 Hz,12H) ,7.70(s,6H),8.58(s,6H);13C NMR(125 MHz,CDCl3):57.4,107.9,120.0,122.8,123.1,128.8,136.1,138.2,142.6,149.1,156.5;元素分析(按C72H54O24S6计算值)/%:N 0.02(0.00),C 57.39(57.83),H 3.55(3.61)。
化合物5和6,向100 mL的两口瓶中加入1.173 g(3 mmol)4,6-二正丙氧基-1,3-苯二磺酰氯2a的二氯甲烷(10 mL)溶液,0.480 g(3 mmol)2,7-二羟基萘的二氯甲烷(10 mL)溶液,再加入0.81 mL(6 mmol)三乙胺,油浴65 ℃回流30 h。 TLC跟踪原料点消失,结束反应。 蒸出溶剂,得粗产品。 上硅胶柱层析,以 V(石油醚)∶ V(乙酸乙酯)=1∶5的混合溶剂洗脱后重结晶干燥,得白色晶体5[16]和6。
化合物5,白色晶体0.26 g,产率18.0%,mp 220~221 ℃;IR(KBr), σ/cm-1:1058,1097,1148,1196,1464,1556,1596;MS(MALDI-TOF) m/z:979.2(M+Na+),995.1(M+K+)(C44H44O16S4,FW 956);1H NMR(500 MHz,DMSO-d6), δ:1.10(t, J=7.2 Hz,12H),1.89~.93(m,8H),4.52(t, J=5.3 Hz,8H),6.63(d, J=7.8 Hz,4H),7.28(s,2H),7.41(s,2H),7.85(s,4H),8.02(d , J=9.0 Hz,4H);13C NMR(125 MHz,DMSO-d6), δ:10.9,22.3,72.6,100.9,113.6,121.4,122.0,130.2,133.8,135.1,147.3,164.1;元素分析(按C44H44O16S4计算值)/%:N 0.04(0.00),C 54.71(55.23),H 4.55(4.60)。
化合物6,白色晶体0.17 g,产率为12.0%,mp 244~245 ℃;IR(KBr), σ/cm-1:1062,1125,1152,1179,1190,1510,1557,1595,1633,2881,2969;MS(MALDI-TOF) m/z:1935.2(M+Na+),1951.2(M+K+)(C88H88O32S8,FW 1912);1H NMR(500 MHz,DMSO-d6), δ:0.93(t, J=7.2 Hz,24H),1.67~1.70(m,16H),4.30(t, J=5.3 Hz,16H),7.02(d, J=9.0 Hz,8H),7. 07(s,4H),7.60(d, J=9.0 Hz,8H),7.72(s,8H),7.83(s,4H);13C NMR(125 MHz,DMSO-d6), δ:10.6,22.0,72.5,100.5,114.1,120.2,121.4,130.3,133.9,134.8,147.8,164.0;元素分析(按C88H88O32S8·8H2O计算值)/%:N 0.07(0.00),C 51.48(51.36),H 5.26(5.06)。
通过合成实验,发现了制备一系列结构对称的芳香磺酸酯桥联大环化合物的方法:由芳香二磺酰氯和芳香二酚反应得到,因此它们含有偶数个芳香环围成的空穴。 通过让不同的磺酰氯和不同的二酚反应建构不同空穴大小的磺酸酯桥联大环3~6,其MALDI-TOF谱图中都出现了分子加一个Na+的峰,荷质比分别为867.3、1516.7、979.2和1935.2,与预期吻合。
磺酸酯桥联大环化合物的成环反应受反应物自身性质、溶剂和温度的影响。
如果反应物溶于二氯甲烷溶剂的话,反应的成功率较高。 一般情况,成环反应会生成四、六、八聚体……,以四聚体和六聚体为主,不同的反应中不同的多聚体的比例不同。 例如Scheme 2所示,化合物7的合成通过MALDI-TOF图谱证明,生成了6种产物,并且随着环的增大,聚合物的含量减少。 由于目标产物薄层色谱分析点板不是一个圆点,而是拖尾到基线,多数反应很难拿到多种产物,一般柱层析后重结晶拿到的是主要产物。 在一些反应中,根据产物溶解性、熔点和柱色谱不同位置,分离得到几种不同的产物。
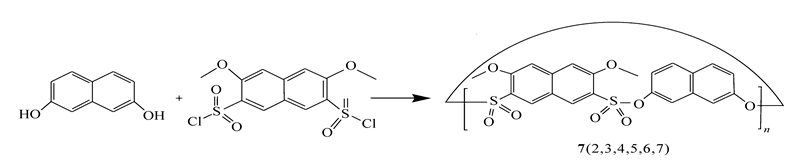 | Scheme 2 Synthesis of the sulfonate-bridged macrocyclic compounds 7 |
{kind=link}
成环反应对溶剂是有选择的,例如水、乙晴、二甲基甲酰胺(DMF)、吡啶、二甲基亚砜(DMSO)、乙醚、丙酮和苯等不易成环。 三氯甲烷作溶剂可以发生环化反应,但产率较低,选用二氯甲烷作溶剂,环化反应产率相对较高。 另外,缚酸剂的选择也是三乙胺最合适,而四丁基碘化铵、碳酸钾和NaH均不适合成环反应。
温度的选择也很重要,反应温度过低,外部提供的能量不足以引发反应,达到活化状态;温度过高由于酚类物质和磺酰氯均容易变质,因而会使反应产率降低,合适的温度是60.0 ℃左右。
在Scheme 1中,Equation c中芳环上的取代基是正丙氧基,Equation a和Equation b中芳环上的取代基是甲氧基,因此产物5和6比3和4的极性小,熔点低。 洗脱时,Equation c所用的混合溶剂为石油醚和乙酸乙酯体系,而反应a和b所用的混合溶剂为甲醇和乙酸乙酯体系。
为了找到温度和环化反应的产率及产物关系的规律性,对生成5和6的反应(如Equation c所示)测试了4个温度,分别为0、27.0、58.0和76.5 ℃。 发现前两个温度此反应不发生,在58.0 ℃时,四聚物5和八聚物6的产率分别是18.0%和12.0%,物质的量比为3∶1;在76.5 ℃时,四聚物5和八聚物6的产率分别是8.6%和11.4%,物质的量比为3∶2。 可见,温度升高,副反应更容易发生,总产率降低。 但随着温度提高,八聚物的相对比例增加,也就是更有利于形成更大环产物。
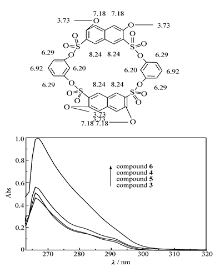
由图1变温核磁图谱数据,化合物6苯环烷氧基支链上的氢化学位移基本没有随温度的变化而变化,说明温度变化过程中带支链的苯环没有翻转。 因为如果翻转的话,当支链进入空腔内的时候,支链氢化学位移要向高场移动。 由此可知,在苯环上聚有支链有利于固定构象,可以通过这种方法合成结构固定的杯芳烃[10,14]。
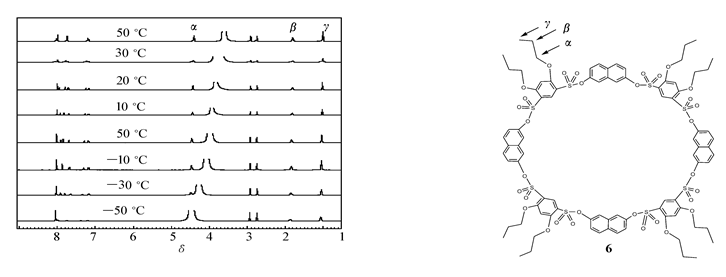 | 图1 化合物6的变温核磁图Fig.1 1H NMR spectra of compound 6 in different temperatures(500 MHz,DMF- d7) |
{kind=link}
目标化合物的结构用IR、1H NMR和MS等进行了确证,以化合物3为例说明如下:化合物3的IR谱中,3109 cm-1较弱的吸收峰是芳环的C—H键伸缩振动谱带;2852~2922 cm-1出现的多个较弱的吸收峰是甲基中C—H键的伸缩振动谱带;1596、1491和1461 cm-1是芳环的骨架伸缩振动谱带;1250和1379 cm-1是 
溶液配制:以DMF为溶剂,配制1.0×10-4 mol/L的化合物3~6溶液,取浓度为1.0×10-4 mol/L的化合物3~6溶液1 mL,用DMF定容至10 mL,取3.0 mL于比色皿中(石英比色皿厚度为1 cm),测定紫外光谱(如图2所示)。
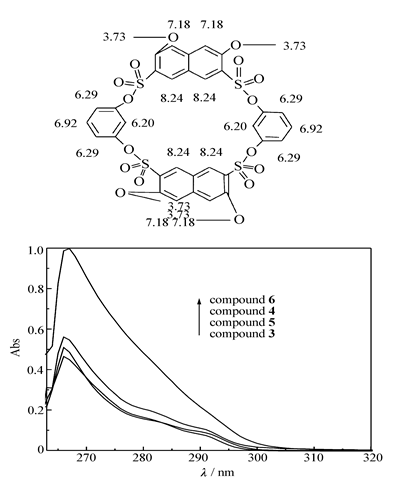 | 图2 化合物3~6在DMF中的紫外吸收光谱Fig.2 UV spectra of compound 3~6 in DMF solutions c(3~6)=1.0×10-5 mol/L |
{kind=link}
化合物3~5的DMF溶液的最大紫外吸收峰在266 nm处,是芳环的π→π*跃迁和磺酸酯基n→π*所产生的吸收带,摩尔吸光系数分别是4.65×104、5.61×104和5.09×104 L/(mol·cm);化合物6的DMF溶液的最大紫外吸收峰在267 nm处,摩尔吸光系数是9.98×104 L/(mol·cm)。
分光光度法中规定扣除空白值后,吸光度为0.01时对应的浓度值为检出限。 此处空白值是指DMF在266和267 nm处的吸光度,可在仪器上扣除,所测得的数据即为样品的吸光度。 因此,根据朗伯-比耳定律,得出化合物3~6的检出限分别是2.15×10-7、1.78×10-7、1.96×10-7和1.00×10-7 mol/L。
以一步环化法合成了一系列环大小不同的磺酸酯桥联大环化合物,产率较高,生成的大环对称性和脂溶性较好,易溶于丙酮中,柱色谱纯化后再重结晶纯化方便可行,同时对这类化合物的合成制备方法进行了有价值的探讨。 这些大环化合物的结构得到了核磁、红外和MALDI-TOF的确认。 变温核磁数据证明,在苯环上加上支链有利于固定构象,可以通过这种方法合成刚性芳香大环化合物[10,14]。 紫外光谱最大吸收峰在266和267 nm处,根据朗伯-比耳定律,计算得化合物3~6的摩尔吸光系数和检出限。 从我们前面研究的结果[12,16]可以预见,磺酸酯类桥联大环化合物与金属配位的良好性能会吸引更多的人的关注,将来会有更好的发展空间。
致谢 感谢为本研究提供指导和帮助的北京师范大学化学学院的龚兵教授和何兰教授。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|
| [14] |
|
| [15] |
|
| [16] |
|