
XIA Ran, SUN Liping, YANG Xining, et al. Metal-free Synthesis of 2,6-Dichloropurineside and 2-Chloroadenosine[J]. Chinese Journal of Applied Chemistry, 32(12): 1398-1401


共同通讯联系人:渠桂荣,教授; Tel/Fax:0373-3329276; E-mail:quguir@sina.com; 研究方向:核苷药物的合成
提出了合成2,6-二氯嘌呤核苷和2-氯腺苷的新方法。 以商品化的2,6-二氯嘌呤和四乙酰核糖为原料,在5%(摩尔分数)三氟甲磺酸催化下,得到缩合物2',3',5'-三- O-乙酰基-2,6-二氯嘌呤核苷。 缩合物在浓H2SO4催化下,以89%的收率得到2,6-二氯嘌呤核苷;在NH3/CH3OH体系中氨解和脱除乙酰基,以92%的收率得到2-氯腺苷。 反应规模可以扩大到100 g,收率未降低。 该方法原料价格低廉,避免使用重金属催化剂,操作简便,中间体及产物可以通过结晶的方法纯化得到,显示出潜在的应用价值。
Co-corresponding author:QU Guirong, professor; Tel/Fax:0373-3329276; E-mail:quguir@sina.com; Research interests:synthesis of nucleoside drugs
New methods for the synthesis of 2,6-dichloropurineside and 2-chloroadenosine were developed. The key intermediate 2',3',5'-tri- O-acetyl-2,6-dichloropurineside was obtained from the condensation of 2,6-dichloropurine and β-D-ribofuranose 1,2,3,5-tetraacetate under the catalysis of 5% molar fraction of trifluoromethanesulfonic acid. 2,6-Dichloropurineside was obtained by sulfuric acid catalyzed hydrolysis of 2',3',5'-tri- O-acetyl-2,6-dichloropurineside in the yield of 89% and 2-chloroadenosine was obtained from the aminolysis of 2',3',5'-tri- O-acetyl-2,6-dichloropurineside in NH3/CH3OH with the yield of 92%. The starting substrates were all commercially available and affordable. The presented method avoided toxic metal catalysts and chromatography. Moreover, 2-thioadenosine was obtained in reliable yield on a 100 g scale.
2位氯代的嘌呤核苷具有显著的抗肿瘤及抗病毒药物活性,其中,2,6-二氯嘌呤核苷(1)对治疗由支原体引起的感染具有较好的疗效[1]。 2-氯腺苷(2)是一种具有多种药理活性的药物[2,3,4],主要用于治疗肺癌、肝癌、乳腺癌和血癌。 它还用于治疗癫痫病、心肌梗塞、原发性高血压等病,治疗效果好,毒副作用小,是目前在国际上使用频率较高的核苷类抗癌、抗病毒药物。 2,6-二氯嘌呤核苷还可以用于合成2-氯腺苷、腺苷A3受体激动剂[5]等。 2-氯腺苷被用来合成临床上使用的核苷类抗白血病药物克拉屈滨[6]和氯法拉滨[7]。 故2,6-二氯嘌呤核苷和2-氯腺苷的合成具有重要的意义。
目前,文献报道的2,6-二氯嘌呤核苷和2-氯腺苷的合成方法中,最大的缺点是在缩合步骤中用到重金属催化剂,如HgCl2[8] 或SnCl4[9] ,毒性大、易残留、环境污染大,合成效率不高,后处理繁琐,限制了进一步的应用。
我们在研究中[10,11]发现,在熔融状态下,加入5%摩尔分数的三氟甲磺酸,四乙酰核糖(3)和2,6-二氯嘌呤(4)能够顺利缩合,只在2,6-二氯嘌呤的N9位生成C—N键,缩合产物经过脱保护得到2,6-二氯嘌呤核苷,通过氨解得到2-氯腺苷。 与文献[8,9]相比,本文的方法避免了使用重金属催化剂,环境友好,操作更为方便,为2,6-二氯嘌呤核苷、2-氯腺苷及其类似物的合成提供了有效的途径(Scheme 1)。
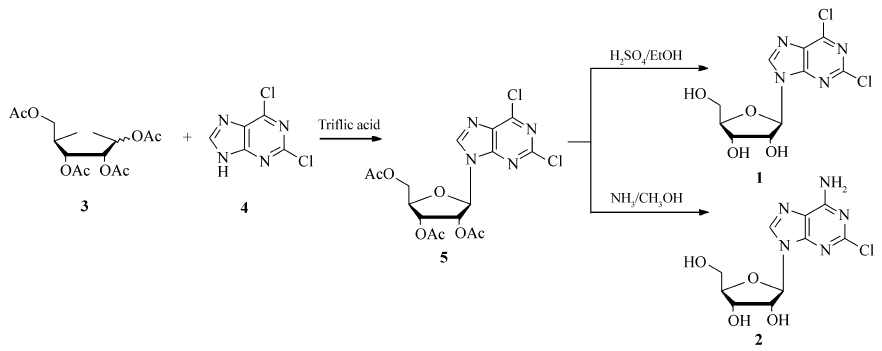 | Scheme 1 The synthetic route of 2,6-dichloropurineside(1) and 2-chloroadenosine(2) |
{kind=link}
AC 400型核磁共振仪(德国Bruker公司),DMSO-d6或CDCl3为溶剂,TMS为内标;Q-TofMS/MS型高分辨质谱(美国Waters公司);XRC-1型显微熔点仪(四川大学科仪厂,温度计未校正)。
四乙酰核糖、2,6-二氯嘌呤(纯度>99%,新乡拓新生化股份有限公司)。 所用试剂均为市售分析纯。 POCl3需要重蒸,CH2Cl2和亚硝酸叔丁酯用4A分子筛干燥处理24 h。 其它试剂未经进一步处理。
1.2.1 2',3',5'-三- O-乙酰基-2,6-二氯嘌呤核苷的合成(5) 四乙酰核糖(1.00 g,3.14 mmol),加热到120 ℃至熔化,加入2,6-二氯嘌呤(0.59 g,3.14 mmol),搅拌,加入催化剂三氟甲磺酸(0.02 mL,0.16 mmol),在真空度0.09 MPa下减压反应1 h,降至室温,反应物逐渐凝固,加入乙醇(10 mL)重结晶,得到2',3',5'-三- O-乙酰基-2,6-二氯嘌呤核苷,1.29 g,收率91%。
四乙酰核糖(100 g,314 mmol),加热到120 ℃至熔化,加入2,6-二氯嘌呤(59 g,314 mmol),搅拌,缓慢加入催化剂三氟甲磺酸(2.40 mL,15.70 mmol),在真空度0.09 MPa下减压蒸出生成的醋酸,反应5 h,降至室温,反应物逐渐凝固,加入乙醇(200 mL)重结晶,得到2',3',5'-三- O-乙酰基-2,6-二氯嘌呤核苷,134.74 g,收率96%。
白色固体,mp 162~164 ℃(文献[12] mp 162~163 ℃)。1H NMR(CDCl3,400 MHz), δ:8.26(s,1H),6.21(d, J=5.6 Hz,1H),5.78(t, J=5.6 Hz,1H),5.56(t, J=4.8 Hz,1H),4.44~4.35(m,3H),2.11(s,3H),2.08(s,3H),2.03(s,3H);13C NMR(CDCl3,100 MHz), δ:170.3,169.6,169.4,154.7,152.7,150.6,144.2,133.6,86.1,80.6,73.1,70.5,62.9,20.8,20.5,20.4;HRMS计算值C16H16Cl2N4NaO7 [M+Na+] 469.0288,实测值469.0288。
1.2.2 2,6-二氯嘌呤核苷的合成(1) 2',3',5'-三- O-乙酰基-2,6-二氯嘌呤核苷(10 g,22.37 mmol),加入到无水乙醇(100 mL)中,慢慢滴加浓H2SO4(5 mL),室温反应10 h,降温,用无水Na2CO3中和,过滤,母液减压浓缩,得到油状物,用90%EtOH重结晶,得到2,6-二氯嘌呤核苷,6.39 g,收率89%。
白色固体,mp 152~154 ℃(文献[9] 152~154 ℃)。1H NMR(DMSO-d6,400 MHz), δ:8.36(s,1H),5.81(d, J=6.0 Hz,1H),5.46(d, J=6.0 Hz,1H),5.19(d, J=4.4 Hz,1H),5.05(t, J=4.8 Hz,1H),4.53~4.48(m,1H),4.12(d, J=4.8 Hz,1H),3.94(d, J=2.8 Hz,1H),3.67~3.51(m,2H);13C NMR(DMSO-d6,100 MHz), δ:156.6,152.9,149.5,140.4,119.8,88.4,86.4,73.9,71.1,62.2;HRMS计算值C10H11Cl2N4O4 [M+H+] 321.0152,实测值321.0152。
1.2.3 2-氯腺苷的合成(2) 2',3',5'-三- O-乙酰基-2,6-二氯嘌呤核苷(10 g,22.37 mmol),加入到饱和的NH3/CH3OH溶液(100 mL)中,加热到50 ℃,保温反应10 h,降温,减压除去溶剂,得到油状物,用90%EtOH重结晶,得到2-氯腺苷,6.21 g,收率92%。
白色固体,mp 154~156 ℃(文献[13] mp 155 ℃)。1H NMR(DMSO-d6,400 MHz), δ:8.37(s,1H),7.83(brs,2H),5.81(d, J=6.0 Hz,1H),5.47(d, J=6.0 Hz,1H),5.19(d, J=4.8 Hz,1H),5.05(t, J=4.8 Hz,1H),4.52~4.48(m,1H),4.12~4.09(m,1H),3.93(d, J=3.6 Hz,1H),3.67~3.62(m,1H),3.56~3.51;13C NMR(DMSO-d6,100 MHz), δ:156.3,152.9,149.8,140.8,118.7,84.5,84.0,76.1,75.4,61.3;HRMS计算值C10H13ClN5O4 [M+H+] 302.0651,理论值302.0658。
以1,2,3,5-四- O-乙酰基- β-D-核糖和2,6-二氯嘌呤为反应底物,考察了催化剂、反应温度和反应时间对缩合反应的影响,结果见表1。
| 表1 反应条件优化 a Table 1 Optimization of reaction conditions a. |
以摩尔分数10%的SnCl4为催化剂,在100 ℃反应1 h,反应没有发生。 当提高温度至120 ℃,反应1 h,收率可以达到85%。 当不加催化剂时,在同样的条件下,反应没有发生,说明文献[9]报道的SnCl4能有效促进反应的发生。 为了筛选出非金属催化剂,考察了一系列有机酸作为催化剂。当使用对甲苯磺酸、三氟乙酸时,收率达到76%和89%。 三氟甲磺酸能够进一步提高收率,达到91%。 当将催化剂的量降低至5%时,收率没有降低。 当继续降低至2%时,收率降低至69%,说明最佳的催化剂投加量为5%。 当反应温度升高至150 ℃时,反应物开始有部分碳化,造成收率降低至78%。 最后,我们考察了反应时间对收率的影响,结果显示,当反应时间为0.5 h时,收率为74%,时间为2 h时,收率90%,所以最佳的反应时间为1 h。 需要指出的是,当反应规模扩大到100 g时,反应时间需要增加到5 h,收率96%,而反应1 h时,收率仅63%,说明反应规模扩大需要更长的反应时间。
常用的脱除乙酰基的体系有NH3/CH3OH、NaOCH3/CH3OH等,这些条件均为碱性环境。 因为嘌呤6位的氯原子非常活泼,在脱除乙酰基的同时,6位的氯原子会被—NH2或CH3O—取代,生成副产物,所以,这些方法不适合2,6-二氯嘌呤核苷的合成。 经过探索发现,酸性条件既能脱除乙酰基,又能防止6位氯原子的取代反应。 最终,以浓H2SO4为催化剂,以无水EtOH为反应溶剂。
在2-氯腺苷的合成中,NH3/CH3OH体系既能脱除乙酰基,又能使6-Cl氨解。 2-Cl性质比6-Cl稳定,即使在加热的条件下,也不会发生氨解反应。
我们发展了一种有效合成2,6-二氯嘌呤核苷和2-氯腺苷的新方法。 以廉价的、商品化的2,6-二氯嘌呤和四乙酰核糖为原料,在5%三氟甲磺酸催化下,在熔融状态发生缩合反应,得到缩合物2',3',5'-三- O-乙酰基-2,6-二氯嘌呤核苷。 缩合物在浓H2SO4催化下,以无水EtOH为反应溶剂,以89%的收率得到2,6-二氯嘌呤核苷;在NH3/CH3OH体系中氨解和脱除乙酰基,以92%的收率得到2-氯腺苷。 缩合反应可以扩大到100 g的反应规模,收率没有降低。 对比传统方法,该方法避免使用重金属催化剂,操作简便,中间体及产物可以通过结晶的方法纯化得到,非常适合于实验室内对2,6-二氯嘌呤核苷和2-氯腺苷的合成,具有潜在的应用价值。
| [1] |
|
| [2] |
|
| [3] |
|
| [4] |
|
| [5] |
|
| [6] |
|
| [7] |
|
| [8] |
|
| [9] |
|
| [10] |
|
| [11] |
|
| [12] |
|
| [13] |
|